
RQM+ Medical Device and In Vitro Diagnostic Blog
Sorry, no listings found for that Search. Try changing your fiter and search again.
-

New FDA Dataset Marks a Transformative Leap for Chemical Characterization
on 23 April 2024 | By Taryn Meade, Jordi Labs/RQM+ Director of Biological Evaluation
We're thrilled to announce a significant development from the FDA’s Center for Devices and Radiological Health (CDRH) that introduces a new regulatory science tool for chemical characterization. This groundbreaking tool is a huge step moving toward improved chemical...
Read More -

Nancy Morrison's Keys to Thriving in Regulatory Affairs: Lessons Learned from a 30+ Year Career
on 11 April 2024 | By Stephen Biernacki, Marketing Principal
In this heartfelt and insightful video presentation, Nancy Morrison – as highly a respected professional as you'll ever find in regulatory affairs – shares her journey and the lessons she's learned along the way. As she embarks on her well-deserved retirement, Nancy generously...
Read More -

Navigating PFAS Regulations: Lab Analysis for Medical Device Material Changes
on 2 April 2024 | By Kevin Rowland, Jordi Labs/RQM+ Director of R&D
Regulations in both the EU and US are rapidly changing in order to address the health and environmental risks posed by per- and poly-fluoroalkyl substances (PFAS). Through the EU REACH regulation, it has been proposed to restrict PFAS which will preclude manufacturers from...
Read More -

Brought to You by the Letter M: Operational Considerations for Transitioning From the QSR to the QMSR
on 20 March 2024 | By Ed Ball, CEng, MIPEM, MIMMM – Manager, Intelligence & Strategic Execution
We used the publication of the final rule amending the US Quality System Regulations (QSR) as an example in our recent blog titled Enhancing Competitiveness in MedTech: Smart Strategies with Regulatory Intelligence. Here we will look at the final rule, and its implications in a...
Read More -

What are Extractables and Leachables?
on 15 March 2024 | By Kevin Rowland, Jordi Labs/RQM+ Director of R&D
Extractables and leachables are potentially hazardous chemicals that a patient can be exposed to when using medical devices or drug products. These chemicals may be present as impurities in the materials used, but also may be introduced during manufacturing or storage....
Read More -

8 Strategic Advantages of Outsourcing Audits
on 14 March 2024 | By Richard Freeman, Director, Global Audit Practice
Staying ahead of compliance and regulatory requirements needs to be a top priority for MedTech companies. Audits are an integral part of maintaining these standards, but can often be seen as stressful and time-consuming events. However, the perspective shifts when these are...
Read More -

Streamlining AE Reporting in PMCF Studies (FDA Perspective)
on 11 March 2024 | By Niki Spaniel, RAC, Principal, CPMP, Practice Manager and Anastassia Young, MS, Senior Consultant
The FDA Exemptions, Variances, and Alternative Forms of Adverse Event Reporting for Medical Devices program may benefit manufacturers in reducing the burden of medical device reporting (21 CFR Part 803, under 21 CFR 80.309(b) where real world data/evidence is being collected,...
Read More -

Classification for SARS-CoV-2: Adjusting to a Post-Pandemic Reality Under IVDR
on 29 February 2024 | By Margot Borgel, Director, IVD Global Regulatory Affairs
Team-NB recently released a position paper regarding the classification of devices to detect SARS-CoV-2. These devices are currently cited in the MDCG 2020-16 classification guidance as an example of a device falling under Rule 1, 2nd indent, which states: "Devices intended to...
Read More -

Checklist for Navigating PFAS Phase-Out in MedTech
on 20 February 2024 | By Jaishankar Kutty, Ph.D., RQM+ VP of Intelligence and Innovation and James Wrenn, Director, Solutions Delivery RQM+
The impending phase-out of per- and polyfluoroalkyl substances (PFAS) presents a significant challenge for MedTech companies. After all, PFAS has been an integral component in manufacturing many MedTech devices.
Read More -

Join Live Webinar: Decoding MDCG 2023-7 for MedTech Devices
on 12 February 2024 | By Amie Smirthwaite BEng, PhD, FRAPS, RQM+ Senior Vice President, Intelligence & Innovation
Published in December last year, MDCG 2023-7 clarifies when clinical investigations are not mandatory for Class III and implantable devices. MDCG 2023-7 also shares updated guidelines for when data from equivalent devices may be used in a clinical evaluation under the EU Medical...
Read More -

Enhancing Competitiveness in MedTech: Smart Strategies with Regulatory Intelligence
on 6 February 2024 | By Ed Ball, CEng, MIPEM, MIMMM – Manager, Intelligence & Strategic Execution
What is regulatory intelligence? Ok, so what on earth are we talking about when we say regulatory intelligence? There are no formal definitions for regulatory intelligence in regulations or standards relating to medical devices or in vitro diagnostic devices. Without delving...
Read More -

The Most Common Issues Affecting Biocompatibility Tests
on 3 January 2024 | By Kevin Rowland, Jordi Labs/RQM+ Director of R&D
While biocompatibility testing is necessary to develop safe MedTech devices, several issues complicate the process. From the initial study setup to deriving accurate conclusions, complications can arise at each testing stage. That’s bad news. Any testing obstacles can delay...
Read More -

Unveiling MDCG 2023-7: Equivalence, Clinical Investigations and Data Access Under EU MDR
on 21 December 2023 | By Amie Smirthwaite BEng, PhD, FRAPS, RQM+ Senior Vice President, Intelligence & Innovation
Introduction to the New MDCG Guidance A new MDCG guidance document has been published today, providing welcome clarification regarding the practical application of EU MDR Articles 61(4) – (6). This new guidance, "MDCG 2023-7: Guidance on exemptions from the requirement to...
Read More -

Preparing for the Phase-Out of PFAS in MedTech Manufacturing
on 12 December 2023 | By Jaishankar Kutty, Ph.D., RQM+ VP of Intelligence and Innovation and James Wrenn, Director, Solutions Delivery RQM+
Major per- and polyfluoroalkyl substances (PFAS) suppliers are beginning to exit the market due to significant environmental, health, regulatory, and legal concerns. That’s mostly a good thing! While PFAS are used in an incredible array of consumer products, including medical...
Read More -

Why You Should Verify Your Supplier's MedTech Materials
on 5 December 2023 | By Kevin Rowland, Jordi Labs/RQM+ Director of R&D
In the MedTech sector, the biocompatibility of materials is critical for patient safety and product efficacy. Every material needs to be tested, with suppliers often providing quality and biocompatibility assurances.
Read More -

Musical Chairs MDR-Style: Keep dancing even though the music has stopped.
on 16 November 2023 | By Ed Ball, CEng, MIPEM, MIMMM – Manager, Intelligence & Strategic Execution
Introduction We have previously talked about the upcoming 2024 EU deadlines for QMS compliance and notified body applications for the EU MDR 2017/745 (5 Essential Tips for EU MDR Compliance and How to Meet the EU's 2024 MDR Deadlines), but what is needed beyond that? What if you...
Read More -

Enough Is Enough: Deciding When Your Safety Evaluation Is Complete
on 10 November 2023 | By Kevin Rowland, Jordi Labs/RQM+ Director of R&D
For any MedTech company, the goal of developing products that are both effective and safe is at the heart of their mission. But as these companies build their products, they inevitably encounter testing challenges. Comprehensive testing, while crucial, often comes with a hefty...
Read More -

How a full-service CRO can get your device to market quicker
on 1 October 2023 | By Eric Pauls, Chief Customer Officer
From navigating the complex regulatory environment to implementing the right quality controls — MedTech companies face many challenges in getting their products to market. These challenges lengthen product development cycles and increase the time it takes to get vital...
Read More -

Don't Get Caught Off Guard: How to Meet the EU's 2024 MDR Deadlines
on 30 August 2023 | By Amie Smirthwaite, Ph.D., RQM+ Senior VP of Intelligence & Innovation and Jaishankar Kutty, Ph.D., RQM+ VP of Intelligence & Innovation
To begin, a cautionary statement... The MDR transition timeline extension is not automatically applied to every legacy device! The timeline extension is conditional in nature, and there are a few key requirements that must be satisfied before manufacturers can benefit from this...
Read More -

5 Tips for IVDR Certification of Companion Diagnostics
on 23 August 2023 | By Margot Borgel, Ph.D.
Introduction The pathway to IVDR certification for companion diagnostics (CDx) can be overwhelming due to the complexity of the certification process. Certification from a notified body was not previously required, as they were self-certified under IVDD. In addition,...
Read More -

Mastering the Transition: 5 Essential Tips for EU MDR Compliance
on 17 July 2023 | By Ed Ball, CEng, MIPEM, MIMMM – Manager, Intelligence & Strategic Execution
Introduction Regulation 2023/607 removed the May 2024 transition deadline for medical devices transitioning from MDD/AIMDD to MDR 2017/745. The end of the transition period was pushed back to 2026, 2027 or 2028, depending on the classification of the device.
Read More -

Breaking New Ground: Introducing the Transitional Coverage for Emerging Technologies (TCET) Pathway
on 5 July 2023 | By Daniel Lace, MD, Chief Medical Officer
This blog post explores the CMS and AHRQ's proposed TCET pathway, which aims to improve beneficiary access, reduce coverage uncertainty, encourage evidence development and coordinate benefit categories for emerging technologies. It also discusses eligibility criteria for...
Read More -

EU MDR Postponed – What We Know Now and What's Next
on 9 December 2022 | By Jaishankar Kutty, Ph.D., RQM+ VP of Intelligence & Innovation
Earlier today, in the EPSCO meeting, all EU member states unanimously agreed to the proposal recommendations listed below. Belgium asked for a detailed root cause analyses of why the MDR roll out finds itself where it is today. Malta has asked for one single date as opposed to a...
Read More -

Summary of FDA Regulatory Changes for Digital Health Devices in Q3 2022
on 6 December 2022 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
Digital health devices are no longer new on the scene, but the regulatory landscape continues to evolve. As predicted in our blog post earlier this year, the latest development is new U.S. Food and Drug Administration (FDA) guidance documents for digital health devices that were...
Read More -

Medical Device Software: Top Regulatory Submission/File Deficiencies and Requests for Additional Info From Both FDA and Notified Bodies
on 29 November 2022 | By Kevin Go, MBA, RAC, CQA, RQM+ Senior Principal
Medical devices that fall into the category of software as a medical device (SaMD) are on the rise as technology continues to evolve. Although these products have proven to be highly beneficial to patients and end users, manufacturers must overcome many regulatory challenges.
Read More -
European Union Expert Panel's Review of a Novel Neurostimulation Device
on 21 November 2022 | By Andrew Tarnaris, M.D. MD (Res) FRCS (NeuroSurg), RQM+ Medical Director
A new opinion (CECP-2022-000222) provided under the Clinical Evaluation Consultation Procedure (CECP) was recently published by the European Commission. Here are some things worth noting, a summary to digest and lessons learnt both for manufacturers and notified bodies alike.
Read More -

How the FDA Encourages Innovation in Pediatric Medical Devices and Best Practices for Submissions
on 8 November 2022 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
As an industry, medical device manufacturers and regulators have an obligation to ensure that children are not left behind when it comes to the innovation and development of products to treat childhood conditions and diseases. The reality is that the innovation of pediatric...
Read More -

Review of RQM+ Findings in Mock Audits and Notified Body Submissions
on 2 November 2022 | By Rem Siekmann, RQM+, Ronald Sills, RQM+
The transition to EU MDR and EU IVDR has come with a steep learning curve for all in the industry, including both manufacturers and notified bodies. With hundreds of MDR and IVDR technical files under our belts, we have learned a lot about the most common findings — and how to...
Read More -

Lessons Learned From Using the FDA Customer Collaboration Portal (CCP) for eCopy and eSTAR
on 3 October 2022 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
As part of its commitment to shift to electronic submission, the Center for Devices and Radiological Health (CDRH) at the Food and Drug Administration (FDA) launched the Customer Collaboration Portal (CCP) in July 2022. The CCP allows medical device manufacturers to...
Read More -

Are in Silico Trials a Fit for the Medical Device Industry?
on 8 September 2022 | By Andrew Tarnaris, M.D. MD (Res) FRCS (NeuroSurg), RQM+ Medical Director
Pappalardo F et al. published a recent paper (in the IEEE JOURNAL OF BIOMEDICAL AND HEALTH INFORMATICS) advocating for a roadmap towards the CE marking of medical devices based on in silico trials. In silico trials simply mean the use of computer modelling and simulation in...
Read More -

RAPS Convergence 2022: A Sneak Peek at What Medical Device Manufacturers Can Expect to Learn from RQM+ Regulatory and Clinical Experts
on 31 August 2022 | By RQM+ Subject Matter Experts
The annual RAPS Convergence is coming soon, and our team is excited to actively contribute to this year’s program with several speaking sessions. We’re covering wide-ranging regulations for medical devices and in vitro diagnostics in the EU and U.S. Here’s a preview of what to...
Read More -
How to Submit FDA CDRH Submissions Online
on 27 July 2022 | By Kevin Go, MBA, RAC, CQA, RQM+ Senior Principal
As part of FDA Center for Devices and Radiological Health's (CDRH) Digital Transformation Initiative, several new tools have been released to strengthen the regulatory submission process. One of these tools is the Customer Collaboration Portal (CCP), which allows users to track...
Read More -

Impact of a High-Quality IVDR Notified Body Application
on 26 July 2022 | By Heike Moehlig-Zuttermeister, PhD, RQM+ VP Intelligence and Innovation, IVD
The transition from IVDD (98/79/EC) to IVDR (EU 2017/746) has been overwhelming for many manufacturers, especially those that have never worked with a notified body because they were previously able to self-certify under the IVDD. Many are now realizing that the IVDR notified...
Read More -

US FDA Update: Is it a Device? After the Fact Design History Files
on 14 June 2022 | By Ashley Clark, MS, RAC (US, Medical Device), RQM+ Principal Specialist and Nancy Morrison, RAC, RQM+ Executive Director, Regulatory & Quality Consulting Services
Summary Genus Medical Technologies upset the US FDA classification schema for imaging agents such as barium sulfate in the landmark litigation ruling1 in the drug turned device manufacturer’s favor that only products meeting the definition of a drug must be regulated as such....
Read More -

Impact of FDA Adoption of ISO 13485
on 7 June 2022 | By Kevin Go, Project Engineer and Nancy Morrison, Executive Director Regulatory and Quality Consulting Services
The FDA’s proposed rule to align its quality system regulation with ISO 13485 has the industry buzzing. What does it mean for medical device and IVD manufacturers? Let’s take a closer look at the proposed changes and what they might mean for manufacturers if they are adopted.
Read More -

3 Tips for Efficiently Preparing Your FDA eSTAR Submission
on 1 June 2022 | By Anike Freeman, RQM+ Principal
As part of the FDA Reauthorization Act (FDARA) and the Medical Device User Fee Amendments (MDUFA IV) of 2017, the U.S. Food and Drug Administration (FDA) committed to developing electronic submission templates. The intent of these templates is to provide guided submission...
Read More -

Overcoming the Most Common Periodic Safety Update Report (PSUR) and Post-Market Surveillance (PMS) Report Challenges
on 18 May 2022 | By Jan Kloiber, Senior Principal Specialist and Celeste Maksim, Chief of Staff, Clinical and Post-Market Services
May 2022 is the one-year milestone for EU MDR and the date of application for IVDR. For medical devices that have already been approved under MDR, annual periodic safety update reports (PSURs) are due for Class IIb and III devices. Since this is the first year and there is still...
Read More -

How Your Medical Director Can Help You Scope SOTA Properly: A Physician's Perspective
on 10 May 2022 | By Andrew Tarnaris, M.D. MD (Res) FRCS (NeuroSurg), RQM+ Medical Director
State of the art (SOTA) is a key element of the technical documentation required for MDR/IVDR certification. As a new requirement for devices in lower-risk classes, many manufacturers are struggling to provide SOTA context that meets notified body expectations. Whether for new...
Read More -

IVDR State of the Art (SOTA)
on 27 April 2022 | By RQM+ Subject Matter Experts
State of the art (SOTA) is a key piece of the IVDR puzzle. Like many of the other required elements, it touches multiple sections of your technical documentation and must be consistently addressed throughout.
Read More -

A Quantitative Approach to Benefit-Risk Determination
on 21 April 2022 | By Bethany Chung, Ph.D., RAC, RQM+ Principal Regulatory Scientist and Jaishankar (Jai) Kutty, Ph.D., RQM+ VP, Intelligence & Innovation
Under the Medical Device Regulation (2017/745) (MDR), demonstrating clinical benefit and quantifying benefit-risk ratios are critical to compliance. A medical device must not be placed on the market if the benefit of the product does not outweigh the risk in a clearly quantified...
Read More -

Risk Management Challenges with IVDR Compliance - This May Be Your Biggest Obstacle
on 13 April 2022 | By Amie Smirthwaite BEng, PhD, FRAPS, RQM+ Senior Vice President, Intelligence & Innovation
IVD manufacturers have had to make significant improvements to their risk files since the days of IVDD. What was once the reality—static and often incomplete files that hardly changed after being placed on the market—is no longer acceptable. However, despite making major headway...
Read More -

What You Need to Know About Significant Changes Under IVDR
on 23 March 2022 | By RQM+ Subject Matter Experts
Until recently, many IVD manufacturers were able to make changes to the design or intended use of their devices with minimal consequences under IVDD (98/79/EC). With the transition to IVDR and the date of application just around the corner, that time window is quickly closing....
Read More -

Using Chemical Characterization to Achieve Biological Equivalence
on 10 March 2022 | By Jaishankar Kutty, Ph.D., RQM+ VP of Intelligence & Innovation
In the EU, a device is considered to be equivalent if and only if it fulfills all the requirements of technical, clinical, and biological equivalence. Of these, biological equivalence tends to be the most nuanced and can be a confusing topic for medical device...
Read More -
How to Prepare for COVID-19 EUA or Enforcement Policy Expiration
on 22 February 2022 | By Ryan Randall, Senior Engineer, RQM+ (former FDA CDRH Lead Reviewer) and Kevin Go, Project Engineer, RQM+ (former FDA CDRH Lead Reviewer)
Since the beginning of the COVID-19 pandemic, CDRH has dealt with an unprecedented workload to ensure timely access to high quality health products that are essential to the nation’s pandemic response. Two years later, the pandemic response remains a top priority as CDRH...
Read More -

Europe: 2021 Year-End Review & Looking Ahead to 2022
on 21 February 2022 | By Amie Smirthwaite BEng, PhD, RQM+ Senior Vice President, Intelligence & Innovation; Nancy Morrison, RQM+ Vice President, Intelligence & Innovation
2021 was another big year in the European regulatory realm as EU MDR and IVDR implementation continued to roll out. Let’s take a look at the highlights and lessons learned that we can apply as we go into 2022.
Read More -

Just Released: MDCG Guidance 2022-3 Verification of Manufactured Class D IVDs by Notified Bodies
on 17 February 2022 | By Heike Moehlig-Zuttermeister, PhD, RQM+ VP Intelligence and Innovation, IVD
The MDCG guidance 2022-3 Verification of manufactured class D IVDs by notified bodies was issued earlier this week on the 15th of February 2022. This guidance is important for manufacturers because it provides a detailed specification of the requirements for the testing plan and...
Read More -

Dr. Heike Moehlig-Zuttermeister Joins RQM+ as IVD Vice President Intelligence and Innovation
on 9 February 2022 | By RQM+ Subject Matter Experts
RQM+ is excited to announce the addition of Dr. Heike Moehlig-Zuttermeister as IVD Vice President Intelligence and Innovation. Heike joins us after nearly eight years at BSI, where she held leadership roles within the IVD team.
Read More -
Software as a Medical Device
on 7 February 2022 | By Brian Hockett, RQM+ and Chad Quistad, RQM+
Advancements in technology over the past several decades have naturally led to advancements in healthcare. Software as a medical device (SaMD) is particularly poised for growth with one of the latest advancements: the Internet of Things (IoT). Because of this and other factors,...
Read More -

EU Update: Expert Panel Views for SARS-CoV-2 Devices
on 2 February 2022 | By Heike Moehlig-Zuttermeister, PhD, RQM+ VP Intelligence and Innovation, IVD
The first three Performance Evaluation Consultation Procedure (PECP) views have been published for a professional SARS-CoV-2 immunoassay and a professional nucleic acid testing (NAT) assay as well as a near-patient testing NAT SARS-CoV-2 product. Considering the actual...
Read More -

In case you missed it: 2021 Expert Content from the RQM+ Knowledge Center
on 31 January 2022 | By RQM+ Subject Matter Experts
“RQM+ is always the first resource I think of when assistance is needed. In addition I cannot state how grateful I am for your organizations thought leadership and resources/content. It’s best in class!” In 2021, we launched the RQM+ Knowledge Center as your one-stop-shop to...
Read More -

FDA CDRH: What will they do in 2022?
on 26 January 2022 | By Ryan Randall, Senior Engineer, RQM+ (former FDA CDRH Lead Reviewer) and Kevin Go, Project Engineer, RQM+ (former FDA CDRH Lead Reviewer)
“It’s tough to make predictions, especially about the future.” Whether baseball Hall of Famer-slash-philosopher Yogi Berra actually said these words or not, the sentiment rings true, as anyone who has tried to predict the course of the COVID-19 pandemic would know. Predicting...
Read More -

Best Practices and Processes for Integrating CERs and Post-Market Surveillance Under EU MDR
on 21 January 2022 | By Jonathan Gimbel, PhD, RQM+ Executive Director, Technical Leadership, Clinical & Post-Market Practice
The EU MDR date of application has come and gone, and most manufacturers are well aware of the new requirements. However, awareness does not necessarily equal effective implementation. One of the main challenges of MDR compliance is creating a strategy that integrates the work...
Read More -

RQM+ Executive Video Briefing: IVDR New Transition Timelines
on 17 January 2022 | By RQM+ Subject Matter Experts
While we are happy and relieved for the IVD industry to have an extension to the IVDR transition period, we’re concerned that the announcement may result in a misperception that there is plenty of time to get the work done.
Read More -

MDR and IVDR Mock Technical Assessments and Design Examinations
on 10 January 2022 | By Amie Smirthwaite BEng, PhD, FRAPS, RQM+ Senior Vice President, Intelligence & Innovation
With the rollout of EU MDR and IVDR, manufacturers are facing new challenges, especially when it comes to technical documentation review. Medical device and IVD manufacturers are accustomed to doing mock audits for quality systems and ISO certification, but these types of...
Read More -

FDA CDRH 2021: A Year in Review
on 5 January 2022 | By Kevin Go, Project Engineer, RQM+ (former FDA CDRH Lead Reviewer) and Ryan Randall, Senior Engineer, RQM+ (former FDA CDRH Lead Reviewer)
Looking back to 2020, as the COVID-19 pandemic gripped the world, much of the FDA’s focus turned to their response to the public health emergency. CDRH in particular reviewed thousands of emergency use authorization (EUA) applications and worked to facilitate the development and...
Read More -

The IVDR Performance Evaluation Report
on 21 December 2021 | By RQM+ Subject Matter Experts
Under the EU In Vitro Diagnostic Medical Devices Regulation 2017/746 (IVDR), every IVD must have a Performance Evaluation Report (PER). PERs consist of three pillars: scientific validity, analytical performance and clinical performance. Collating data to satisfactorily address...
Read More -

EU MDR Article 61 and equivalence – is there a new way forward?
on 20 December 2021 | By Amie Smirthwaite BEng, PhD, FRAPS, RQM+ Senior Vice President, Intelligence & Innovation
About this article: Could there be more scope for use of equivalence data under EU MDR than you may have thought? Dr. Amie Smirthwaite, Sr. VP of Global Intelligence & Innovation at RQM+, EU MDR expert, and member of the working groups that authored MDCG 2020-5 and 2020-6, has...
Read More -

UK Approved Bodies: on your UKCA mark, get set, go!
on 17 December 2021 | By Ed Ball, CEng, MIPEM, MIMMM – Manager, Intelligence & Strategic Execution
The world of medical device regulation has seen immense change in past years. The EU MDR has been at the forefront of every agenda for some time – and the one-year deadline delay provided valuable extra time for businesses to get their act together. That one-year delay also...
Read More -

Clinical Evidence Requirements Under IVDR
on 16 December 2021 | By RQM+ Subject Matter Experts
The next key regulatory milestone for manufacturers with products on the European market is the EU In Vitro Diagnostic Medical Devices Regulation 2017/746 (IVDR). A critical stage of IVDR planning is for manufacturers to assess whether they have enough clinical and performance...
Read More -
What do we know so far about post-Brexit medical device regulation?
on 14 December 2021 | By Ed Ball, CEng, MIPEM, MIMMM – Manager, Intelligence & Strategic Execution
What do we know so far about post-Brexit medical device regulation? With the new European MDR and IVDR regulations at the forefront of every conversation across the medical device industry, the emerging UK regime may have been overlooked as another pending compliance challenge....
Read More -

Canada Regulations News for Medical Devices and IVDs
on 3 December 2021 | By Nancy Morrison, RQM+
In case you missed it, the Medical Device Regulations in Canada quietly got updated in December 2020. The new regulation intends to boost post-market safety and requires manufacturers to submit a summary report, which is similar to the post-market Periodic Benefit-Risk...
Read More -

Impact of of MDCG 2021-24 on the Classification of Spinal Implants
on 30 November 2021 | By Nancy Morrison, RQM+
The recently released guidance on classification of medical devices is very concerning for manufacturers with spinal implantable medical devices. MDCG 2021-24 appears to have classified many spinal fusion devices that were previously thought to be Class IIb as Class III. This...
Read More -

Is Your Medical Device a Well-Established Technology (WET)?
on 17 November 2021 | By Jonathan Gimbel, PhD, RQM+ Executive Director, Technical Leadership, Clinical & Post-Market Practice
As guidance committees, manufacturers, and notified bodies continue to interpret the EU MDR, some areas remain fuzzy. Well-established technology (WET) is high on the list of definitions that are disputed as notified bodies seem to have a different interpretation than the...
Read More -

Overcoming Challenges and Streamlining Your SSCPs and SSPs
on 8 November 2021 | By RQM+ Subject Matter Experts
In case you missed it, the latest presentation in our webinar series covers the new requirements under EU MDR and IVDR: the Summary of Safety and Clinical Performance (SSCP) and Summary of Safety and Performance (SSP), respectively. It’s no surprise that manufacturers are...
Read More -

What Are Clinical Benefits and How Do You Evaluate Them in Your CER?
on 27 October 2021 | By Jonathan Gimbel, PhD, RQM+ Executive Director, Technical Leadership, Clinical & Post-Market Practice
The rollout of the EU MDR has created new challenges for medical device manufacturers, including those that have had devices on the market for a long time. One area of increased focus is the need to quantitatively establish the clinical benefits to patients from the device based...
Read More -

Med Device Monday - Luminopia One, A Digital Therapeutic to Help Treat Amblyopia
on 25 October 2021 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

Facts & FAQs: European Commission Proposal for Amendment of the IVDR (EU) 2017/746
on 19 October 2021 | By RQM+ Subject Matter Experts
Background The COVID-19 pandemic has led to unprecedented challenges for the In Vitro Diagnostic Regulation (IVDR) implementation impacting both manufacturers and key European Union (EU) infrastructure. On one hand, manufacturers were faced with staff shortages impacting their...
Read More -

Integrating Post-Market Surveillance Into Your QMS
on 12 October 2021 | By Amie Smirthwaite BEng, PhD, FRAPS, RQM+ Senior Vice President, Intelligence & Innovation
Now that medical device manufacturers have begun to implement MDR-compliant post-market surveillance (PMS) systems, it is a good time to take stock of common areas of difficulty and inefficiency, and consider how best to address these. Even manufacturers who are continuing to...
Read More -

FDA Friday - Anike Freeman, M.Eng., PMP
on 8 October 2021 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
This #FDAFriday series consists of mini-interviews with former FDA regulators. Our goals are twofold: (1) help students and professionals interested in Regulatory Affairs see what career paths are possible, and (2) talk about some of the various roles at FDA to demonstrate the...
Read More -

Post-Market Surveillance: A Concise Overview of Requirements
on 5 October 2021 | By Celeste Maksim, PhD, RAC, RQM+
Following the May 2021 deadline, medical device manufacturers now have an MDR-compliant post-market surveillance (PMS) system in place. However, there are still many issues and questions around this topic which need addressing.
Read More -

Top 10 Tips on How to Write a PMS Report and PSUR Without an Official Guidance Document
on 27 September 2021 | By Jan Kloiber, RQM+ Principal
Manufacturers have relied on guidance from the Medical Device Coordination Group and similar organizations to interpret many of the new regulations under EU MDR and IVDR. However, guidance has not been provided for every aspect of the regulation, and application deadlines have...
Read More -

Dr. Andrew Tarnaris Joins RQM+ as Medical Director
on 21 September 2021 | By RQM+ Subject Matter Experts
RQM+ is excited to announce the addition of Dr. Andrew Tarnaris as Medical Director. With a background as both a medical doctor and notified body clinical expert, he represents an excellent example of our unique combination of clinical and regulatory experience.
Read More -
EU Update: SARS-CoV-2 In Vitro Diagnostic Medical Device Performance Evaluation MDCG Guidance Published
on 13 September 2021 | By Ashley (Ash) Clark, MS, RAC, RQM+
A new guidance document (MDCG 2020-21) has been published on the 3rd of August by the Medical Device Coordination Group (MDCG) pertaining to performance evaluation of SARS-CoV-2 in vitro diagnostic medical devices. This document provides extensive details on what is expected for...
Read More -

RQM+ MDR Clinical Evaluation Roadmap: An Interview with Dr. Jai Kutty
on 9 September 2021 | By RQM+ Subject Matter Experts
The introduction of MDR caused a major transformation in the medical device industry. Manufacturers continue to grapple with all elements of compliance, but clinical evaluation presents unique challenges. RQM+ is committed to helping clients navigate these challenges with former...
Read More -

Join us at the 2021 RAPS U.S. Convergence
on 30 August 2021 | By Stephen Biernacki
It is that time of year again for RAPS Convergence and we cannot wait to join the largest annual gathering of regulatory professionals from around the world. The conference will be held virtually this year from 12-15 September 2021 and we will be joining in the fun with a Q&A...
Read More -

RQM+ Announces Investment by Linden Capital Partners
on 16 August 2021 | By Lisa Casavant, RQM+ EVP
Investment to Accelerate Growth and Expand Consulting Services for Medical Device and Diagnostics Manufacturers
Read More -

RQM+ Case Study: Effective Managed Outsourcing for EU MDR
on 2 August 2021 | By RQM+ Subject Matter Experts
THE PROJECT A leading global company offering medical technology, services, and solutions engaged RQM+ to help four business units prepare for the EU Medical Device Regulation (MDR). Specifically, RQM+ is focused on assessing and remediating gaps in MDD technical files and...
Read More -

An Overview of Summary of Safety and Performance (SSP) Under IVDR
on 28 June 2021 | By Nancy Morrison, RQM+
EU IVDR 2017/746 includes a requirement to write a summary of safety and performance (SSP) for Class C and D IVDs. This is a new requirement that wasn’t included in IVDD. The goal of this requirement is to enhance transparency and provide adequate public access to summarized...
Read More -
![[Webinar] Strategies for Overcoming the Biggest Challenges in Achieving IVDR Compliance](https://www.rqmplus.com/hs-fs/hubfs/Blog_Images/RQMPlus%20Webinar.jpg?length=450&name=RQMPlus%20Webinar.jpg)
[Webinar] Strategies for Overcoming the Biggest Challenges in Achieving IVDR Compliance
on 21 June 2021 | By RQM+ Subject Matter Experts
The latest addition to our webinar series covers the biggest challenges in IVDR compliance. With hundreds of MDR submissions under our belts and a number of clients who have already received findings from their IVDR applications, we have learned a lot about the regulation, the...
Read More -

-

RQM+ Quick Guide to Medical Device Recall Management
on 10 June 2021 | By Kim Platt
Medical device recalls are daunting in many ways—manufacturers dread them, competitors love them, and patients don’t always understand them. A medical device recall could be a response to patient safety concerns or to unanticipated negative trends. All manufacturers should...
Read More -

How to Prepare a Design History File for FDA Inspection
on 1 June 2021 | By RQM+ Subject Matter Experts
Food and Drug Administration (FDA) inspections are stressful, especially if your documentation isn’t audit-ready. Creating and maintaining a compliant design history file (DHF) will help ensure that when the time comes, you’ll be ready for an FDA inspection that results in...
Read More -

A Comparison of IVDR to FDA IVD Regulatory Submission Requirements
on 27 May 2021 | By Stephanie Martinez
The European Union’s new In Vitro Diagnostic Regulation (IVDR) May 2022 implementation date is fast approaching. If you sell IVDs in multiple markets, it may be challenging to determine the differences between the IVD regulations in the U.S., the existing IVDD, and the new IVDR....
Read More -

The EU MDR Date of Application Has Arrived!
on 26 May 2021 | By Nancy Morrison, RQM+
The European Union (EU) Medical Device Regulation (MDR) 2017/745 date of application is here! To celebrate the incredible effort medical device manufacturers have put in so far to get to this point, we have compiled a brief review of the remaining EU MDR timeline and a list of...
Read More -
Med Device Monday - The Sonalleve MR-HIFU System Approved to Treat Osteoid Osteomas
on 25 May 2021 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

Optimizing your Technical Documentation to reduce NB review time and questions (Medical Devices and IVDs)
on 20 May 2021 | By Nancy Morrison, RQM+
Whether you are used to working with a notified body or the MDR/IVDR requirements no longer allow you to self-certify and it’s all new to you, it is more important than ever to optimize your technical documentation. Notified bodies are already swamped with MDR submissions, and...
Read More -

How to Obtain CE Marking Under the MDR
on 18 May 2021 | By Nancy Morrison, RQM+
In the EU, CE marking is the system used to demonstrate that products meet the region’s requirements for safety, health, and environmental protection. When a product is labeled with the CE marking, it can be freely traded in the European Economic Area (EEA) without restrictions.
Read More -

Join the RQM+ IVDR Session During RAPS Euro Convergence 2021
on 5 May 2021 | By Deven Hennon, RQM+
We are excited to host a panel discussion during next week's virtual RAPS Euro Convergence 2021! Join our expert panel for our session on IVDR Implementation Best Practices and Challenges. See the schedule here and find more details on our session below.
Read More -

Everything You Need to Know About the Medical Device Single Audit Program (MDSAP)
on 28 April 2021 | By Rem Siekmann, RQM+
Maintaining regulatory and quality compliance across multiple markets is challenging. In addition to meeting stringent regulatory requirements, manufacturers must also submit to regular quality system audits. When operating in multiple markets, this often means a busy audit...
Read More -

Quick Guide to Medical Device Standards: ISO Standards and Beyond
on 28 April 2021 | By Rem Siekmann, RQM+
In the regulatory world, standards are essentially recommended processes that have been developed by subject matter experts with the aim of describing the best possible way to meet an end goal. For example, quality management standards are designed to improve efficiencies and...
Read More -

ISO 14971: 2019: 3 Key Changes from ISO 14971: 2007
on 28 April 2021 | By Chad Quistad, RQM+
As the underlying risk management process for medical devices, the ISO 14971 standard is a critical component of regulatory and quality compliance. When new versions are released and harmonized, it’s up to manufacturers to learn about the changes to update systems accordingly....
Read More -

How to Create a Compliant Periodic Safety Update Report (PSUR) Under EU MDR and EU IVDR
on 28 April 2021 | By Celeste Maksim, PhD, RAC, RQM+
The requirement for a periodic safety update report (PSUR) or post-market surveillance report (PMSR) is new under EU MDR and EU IVDR. MDD and IVDD had post-market requirements, but the MDR and IVDR requirements are more formal, and for most classes, these reports must be updated...
Read More -

Top 4 Findings from Notified Body Technical Documentation Audits Under EU MDR
on 28 April 2021 | By Melissa DeHass, RQM+
As we complete regulatory submissions under MDR, we continue to learn from notified body feedback and audit findings. These lessons learned are captured and disseminated throughout the RQM+ team to build our unrivaled collective knowledge, but we also believe in sharing this...
Read More -

How to Obtain CE Marking Under the IVDR
on 28 April 2021 | By Nancy Morrison, RQM+
CE marking is the system used in the EU to indicate that products meet the region’s safety, health, and environmental protection requirements. Products labeled with the CE marking can be freely traded in the European Economic Area without restrictions.
Read More -

8 Lessons Learned After Writing the Summary of Safety and Clinical Performance (SSCP) using MDCG 2019-9
on 28 April 2021 | By Nancy Morrison, RQM+
EU MDR 2017/745 includes a requirement to write a summary of safety and clinical performance (SSCP) for implantable and Class III devices. Although this is still a relatively new requirement, RQM+ has already gained valuable experience preparing SSCPs on behalf of our clients....
Read More -
Med Device Monday - Novel Device to Reduce Snoring and Mild Obstructive Sleep Apnea
on 26 April 2021 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

RAPS Webcast: PMCF User Feedback Surveys
on 8 April 2021 | By Stephen Biernacki
Update: the RAPS + RQM+ webcast is now available on demand here. Understand the ins and outs of PMCF user feedback surveys by registering for two upcoming and free RQM+ webinars: April 27 – PMCF User Feedback Surveys: Practical solutions for leveraging user feedback surveys as...
Read More -

FDA Friday- Misleading Registration Certificates: Sorting through Company Claims
on 19 March 2021 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -
Med Device Monday - FDA Approves Prosthetic Implant for Above-the-Knee Amputations
on 22 February 2021 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

PMCF User Feedback Surveys
on 17 February 2021 | By Stephen Biernacki
In this webinar we will provide practical solutions for leveraging user feedback surveys as PMCF activities under the EU MDR, presented by former notified body leadership and PMCF subject matter experts.
Read More -

MDCG 2020-16 Classification Guidance for IVDs
on 16 February 2021 | By Ashley (Ash) Clark, MS, RAC, RQM+
RQM+ experts summarize the MDCG guidance from November 2020, Guidance on Classification Rules for the in vitro Diagnostic Medical Devices under Regulation (EU) 2017/746, to aid IVD industry stakeholders in understanding, planning, and executing compliance to the IVDR by May...
Read More -

Dr. Jaishankar Kutty Joins RQM+
on 1 February 2021 | By Deven Hennon, RQM+
February 1, 2021 - RQM+ welcomes Dr. Jai Kutty.
Read More -

New Guidance: FDA Safer Technologies Program (STeP) for Medical Devices
on 21 January 2021 | By Kevin Go, MBA, RAC, CQA, RQM+ Senior Principal
U.S. Food & Drug Administration (FDA) released the highly anticipated Safer Technologies Program (STeP) Guidance Document.
Read More -

Best Practices for Scientific Database Searching
on 18 January 2021 | By Stephen Biernacki
This on-demand webinar will address best practices in scientific database search techniques that can be applied in the performance of a clinical evaluation.
Read More -

Announcing RQM+
on 11 January 2021 | By Deven Hennon, R&Q
We are proud to announce that R&Q and Maetrics have merged and rebranded to form the world's leading medical device and diagnostics consultancy. Check out the full press release below or visit our new brand microsite at RQMplus.com
Read More -
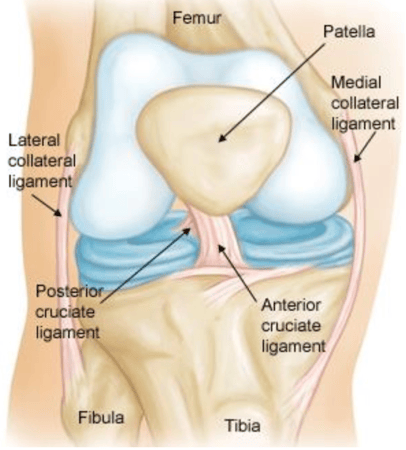
Med Device Monday - New Implant to Repair Torn ACL
on 4 January 2021 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

Leveraging PMCF Surveys for EU MDR Compliance - Part 2
on 15 December 2020 | By RQM+ Subject Matter Experts
When to use Post-Market Clinical Follow-up (PMCF) Surveys under the European Union's Medical Device Regulation.
Read More -
EUDAMED Delay Brings Little Comfort to Medical Device Companies
on 7 December 2020 | By Deven Hennon, R&Q
7 December 2020 Update: After some delays the first module of the European database on medical devices (EUDAMED) is live! Here is what you need to know about the Actor registration module, the first of six modules in EUDAMED: WHO can use this module? Economic operators...
Read More -

Leveraging PMCF Surveys for EU MDR Compliance - Part 1
on 1 December 2020 | By Deven Hennon, R&Q
1 December 2020 - Getting started with Post-Market Clinical Follow-up (PMCF) Surveys under the European Union's Medical Device Regulation.
Read More -

Lessons Learned: 2020 RAPS U.S. and Euro Convergence
on 23 November 2020 | By RQM+ Subject Matter Experts
RQM+ sponsored both RAPS U.S. and Euro Convergence conferences in 2020 and we also led and provided subject matter experts for two panel discussions. Our experts pulled together the highlights from both conferences and summarized them by category for our readers.
Read More -

A New Approach to Corporate Holiday Gift Giving
on 18 November 2020 | By Lisa Casavant, RQM+ EVP
18 November 2020 - A challenging year makes it more important than ever to be supporting and serving our communities.
Read More -

Top Ten Tips and Best Practices for PMCF Surveys Under EU MDR
on 10 November 2020 | By Celeste Maksim, PhD, RAC, RQM+
10 November 2020 - RQM+ experts have come up with a list of our top ten suggestions for mastering Post-Market Clinical Follow-up ("PMCF") Surveys under the European Union's Medical Device Regulation.
Read More -

FDA Friday - Interview with Dr. Seth Carmody
on 30 October 2020 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

FDA’s Tool for Assessing Medical Device Cybersecurity Vulnerabilities
on 28 October 2020 | By Deven Hennon, R&Q
28 October 2020 - October is National Cybersecurity Awareness month, so we think there is no better time to talk about FDA’s new qualified tool for assessing medical device cybersecurity vulnerabilities and why using it makes sense to help you #BeCyberSmart.
Read More -
Med Device Monday - Cybersecure Medical Devices
on 26 October 2020 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

FDA Friday - Cybersecurity Awareness Month at FDA!
on 23 October 2020 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

Q&A: Economic Operators - Addressing the EU MDR and IVDR requirements
on 21 October 2020 | By Nancy Morrison, RQM+
R&Q's August 2020 webinar focused on Economic Operators: Addressing the EU MDR and IVDR requirements head-on. In this follow-up blog post, our knowledgeable MDR and IVDR regulatory experts sat down to answer audience questions.
Read More -

Integrating Risk and Complaint Management
on 16 October 2020 | By Stephen Biernacki
Having strong integrated complaint and risk management processes is about more than just compliance. Watch this webinar and learn critical strategies and tactics for success. As with every R&Q webinar, all registrants will receive access to the slides and recording.
Read More -

Irish Notified Body Requires ISO 14971:2019 Compliance by December 2020
on 14 October 2020 | By Deven Hennon, R&Q
14 October 2020 - The National Standards Authority of Ireland (NSAI) has adopted December 2020 as the expected implementation date for EN ISO 14971:2019.
Read More -

A Discussion With the European Commission on Expert Panels
on 7 October 2020 | By Deven Hennon, R&Q
7 October 2020 - A short conversation with the European Commission provides details on when we can expect access to their expert panels.
Read More -

What You Need to Know About the UK's New Medical Device Guidance
on 1 October 2020 | By Deven Hennon, R&Q
1 October 2020 - As the end of the BREXIT transition period nears, the UK government has released a proposed regime on the regulation of medical devices.
Read More -

R&Q Acquires Maetrics to Form the Largest Medical Device-Focused Global Regulatory and Quality Consultancy
on 10 September 2020 | By Deven Hennon, R&Q
We are proud to announce that we have acquired Maetrics, a leader in life sciences quality and regulatory consulting. Check out the full press release below or visit ourRAPS Convergence 2020 announcement page for more information.
Read More -

Program Management for IVDR and EU MDR
on 8 September 2020 | By Stephen Biernacki
Whether you’re just starting to plan your IVDR program (or not sleeping well because you haven’t started yet) or well into EU MDR but ready to plan post-market surveillance activities, our September webinar will be helpful. All registrants will receive access to the slides and...
Read More -
Announcing R&Q Training Workshops
on 3 September 2020 | By Deven Hennon, R&Q
3 September 2020 - Training Workshops brought to you by the Subject Matter Experts at Regulatory & Quality Solutions.
Read More -

Roundup: RQM+ at the 2020 RAPS Convergence
on 30 August 2020 | By Stephen Biernacki
RQM+ is counting down the days until we can join the RAPS community at the virtual RAPS Convergence 2020 on 13-16 September 2020. As many of you know, the Convergence is the largest annual gathering of regulatory professionals from around the world. While we'll miss interacting...
Read More -

Ranked: The most popular on-demand panel discussions from R&Q
on 24 August 2020 | By Stephen Biernacki
Note: A version of this post was initially published for the flagship online publication of RAPS, Regulatory Focus™ In an effort to further support the medical device industry during the COVID-19 pandemic, we've been spreading love more than ever in one of the best ways we know...
Read More -

R&Q's Ruthanne Vendy Contributes to RAPS Fundamentals of EU Regulatory Affairs
on 11 August 2020 | By Deven Hennon, R&Q
11 August 2020 - We sat down to with R&Q's Ruthanne Vendy, RAC to talk about her experience as a contributing author to the Regulatory Affairs Professionals Society’s (RAPS) Fundamentals of EU Regulatory Affairs, Ninth Edition.
Read More -

Economic Operators: EU MDR and IVDR Requirements
on 3 August 2020 | By Stephen Biernacki
In our August 2020 webinar you'll learn about the EU MDR and IVDR requirements for economic operators.
Read More -

Q&A: Solving the EU MDR Labeling Puzzle
on 29 July 2020 | By Deven Hennon, R&Q
Although a lot has changed in our worlds since R&Q's December 2019 webinar focused on Solving the EU MDR Labeling Puzzle, the general requirements have stayed the same. In this blog post, our knowledgeable EU MDR regulatory experts sat down to provide some updates on a few more...
Read More -

First Do No Harm: Protecting Patients Through Post-Market Surveillance
on 15 July 2020 | By Deven Hennon, R&Q
15 July 2020 - R&Q experts discuss "First Do No Harm - The report of the Independent Medicines and Medical Devices Safety Review."
Read More -
PMCF Plans
on 7 July 2020 | By Stephen Biernacki
In this on-demand webinar originally presented in July 2020, find out how to create detailed, compliant, and business-balanced PMCF plans.
Read More -

Q&A: FDA Emergency Use Authorization (EUA) and EU MDR Article 59
on 6 July 2020 | By Deven Hennon, R&Q
R&Q's May 2020 webinar focused on FDA Emergency Use Authorization (EUA) process and EU MDR Article 59 to release devices needed for prevention and treatment of COVID-19. In this blog post, our knowledgeable FDA and EU MDR regulatory experts sat down to provide answers to your...
Read More -

State of the Art (SOTA) Part I: What is a State of the Art Evaluation?
on 22 June 2020 | By Susan Farabaugh, RQM+ Consultant
You have been reviewing the 4th revision of the European Commission’s Guidelines on Medical Devices, "MEDDEV 2.7/1 Clinical Evaluation: A Guide for Manufacturers and Notified Bodies Under Directives 93/42/EEC and 90/385/EEC" to make sure you are following all requirements to get...
Read More -

Q&A: RAPS Webcast - PMS Requirements of the EU MDR
on 15 June 2020 | By Deven Hennon, R&Q
R&Q is a Premium Solutions Partner of the Regulatory Affairs Professionals Society (RAPS) and in April, we joined forces to offer up a premium webcast. R&Q Executive Director of Regulatory and Quality Consulting Services, Nancy Morrison, presented PMS Requirements of the EU MDR:...
Read More -

Q&A: Structuring PERs under IVDR
on 8 June 2020 | By Deven Hennon, R&Q
R&Q's April 2020 webinar focused on Structuring Performance Evaluation Reports (PERs) under In Vitro Diagnostic Regulation (IVDR). In this blog post, our knowledgeable IVDR experts sat down to provide answers to your questions from the webinar and we hope that they will shed...
Read More -

Emergency Use Authorization Stories: The Humn Project
on 18 May 2020 | By Deven Hennon, R&Q
As we collectively watch the world struggle to cope with the devastating impacts of COVID-19, the Emergency Use Authorization Act has given the R&Q team opportunities to provide pro bono regulatory guidance to incredible medical device companies that are prioritizing “giving...
Read More -

FDAnews Webinar featuring R&Q's Jon Gimbel, Ph.D. on June 2
on 13 May 2020 | By Deven Hennon, R&Q
Tuesday 2 June 2020 — An FDAnews Webinar featuring R&Q
Read More -

Case Studies of MDD and MDR Audit Findings... and Lessons Learned
on 8 May 2020 | By Stephen Biernacki
On-Demand - A webinar entirely dedicated to case studies.
Read More -

RQM+ Remediation Station: Tools & Tips
on 8 May 2020 | By RQM+ Subject Matter Experts
Welcome to the Remediation Station! We've got live shows with the experts, case studies, and insights to help you get through remediation successfully.
Read More -

RQM+ Live! #5 — 5/14/20
on 1 May 2020 | By Stephen Biernacki
Chatting with Former FDA and Notified Body Representatives
Read More -

R&Q Experts Reviewed the New MDCG Guidance - Here is What They Learned.
on 1 May 2020 | By Deven Snyder
If you were excited when the European Commission finally released four guidance documents covering Clinical Evaluation Equivalence, Clinical Evidence of Legacy Devices, and templates for the Post-market clinical follow-up (PMCF) Evaluation Reports and Plans (MDCG 2020-5-8) last...
Read More -

Q&A: EU MDR Audits: Preparing, Managing & Responding to Nonconformances
on 28 April 2020 | By Deven Snyder
R&Q's March 2020 webinar focused on dealing with EU MDR Audit Nonconformances. In this blog post, our knowledgeable EU MDR experts sat down to provide answers to your questions from the webinar and we hope that they will shed some light on similar questions that you may have on...
Read More -
FDA Emergency Use Authorization (EUA) and EU MDR Article 59
on 27 April 2020 | By Stephen Biernacki
26 May 2020 - Processes, tips, and lessons learned.
Read More -

What is Dynamic Mechanical Analysis Used For?
on 24 April 2020 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
Dynamic mechanical analysis, sometimes abbreviated to DMA, is a workhorse technique used to investigate the various mechanical properties of materials as a function of frequency, temperature, and time. The goal is to ultimately get an in-depth assessment of the stress and strain...
Read More -

BREAKING NEWS: European Parliament Approves One Year EU MDR Delay
on 17 April 2020 | By Deven Snyder
This post is an update to our recent blog post where Nancy Morrison, R&Q's Executive Director, Regulatory & Quality Consulting Services, shared her thoughts on the potential impact of the COVID-19 pandemic on the EU MDR timeline. You can read that post here.
Read More -

Focusing on Process Improvements as a Competitive Advantage
on 15 April 2020 | By Deven Snyder
At R&Q, we are seeing firsthand how heavily impacted companies of all sizes are by the volatile global markets brought on by the COVID-19 pandemic. To assist our country’s more vulnerable businesses, the Coronavirus Aid, Relief, and Economic Security (CARES) Act was signed in...
Read More -

COVID-19 Continues to Impact Medical Device Manufacturing in China
on 13 April 2020 | By Deven Snyder
Medical device companies often have a globally distributed workforce which can include remote specification developers and, more likely, manufacturing sites. In China alone, where the virus was first detected, many medical device companies have manufacturing sites, utilize...
Read More -

Join Us Weekly — Announcing DEVICE L❤️VE Live!
on 9 April 2020 | By Stephen Biernacki
The current pandemic is challenging for everyone to varying degrees. R&Q has the utmost respect, appreciation, and love for healthcare workers and all frontline workers across the globe. Our mission from the start has been to improve people's lives and our announcement today has...
Read More -
RAPS Webcast: PMS Requirements of the EU MDR
on 6 April 2020 | By Stephen Biernacki
Note: this webcast is now available on demand. R&Q is a Premium Solutions Partner of the Regulatory Affairs Professionals Society (RAPS) and on Wednesday, April 15th, we're joining forces to offer up a premium live webcast. Attendees will earn 1.5 RAC credits and the event is...
Read More -

Q&A: PMS Requirements of the EU MDR
on 3 April 2020 | By Stephen Biernacki
In February – just before COVID-19 began to seriously impact those of us in the United States – R&Q Executive Director of Regulatory & Quality Consulting Services, Nancy Morrison, presented the webinar PMS Requirements of the EU MDR: Implementation Challenges and Solutions. We...
Read More -

Structuring PERs under IVDR
on 1 April 2020 | By Stephen Biernacki
Whether you are interested in improved strategies for completing PERs or effectively completing your first PER, our April 2020 webinar can act as a critical guide.
Read More -

How Will COVID-19 Impact the EU MDR Timeline?
on 19 March 2020 | By Deven Snyder
Nancy Morrison, R&Q's Executive Director, Regulatory & Quality Consulting Services, shares her thoughts on the potential impact of the COVID-19 pandemic on the EU MDR timeline: Team NB, the European Association of Notified Bodies (NB) that are active in the medical device...
Read More -

New EU MDR Guidance on Significant Changes
on 18 March 2020 | By Deven Snyder
As most of the world seems to be at a standstill this week, the European Commission published new guidance (MDCG 2020-3) to further define “significant change” requirements under the new Medical Device Regulation (MDR) policies coming our way on May 26, 2020. The highly...
Read More -

EU MDR Audits
on 5 March 2020 | By Stephen Biernacki
Preparing, managing and responding to nonconformances.
Read More -

Q&A: Developing an FDA Regulatory Strategy
on 28 February 2020 | By Stephen Biernacki
R&Q's first webinar of 2020 was the first in a two-part series on bringing your medical device to market in the United States. Part one addressed Class I and Class II devices, while part two (TBA in the spring) will cover Class III devices. The questions we received related to...
Read More -

EU MDR 2017/745 Health Check List
on 24 February 2020 | By Deven Snyder
Check out R&Q experts' European Union Medical Device Regulation (MDR) 2017/745 Health Check List below to determine the areas to focus your resources in the final few months before the May 26, 2020 MDR Date of Application. If you have questions, concerns, or need any help...
Read More -

Working Principles and Applications of Scanning Electron Microscopy
on 17 February 2020 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
Scanning electron microscopy (SEM) is an advanced analytical tool that vastly outstrips the capabilities of traditional light microscopy. The standard array of magnifying lenses in a compound microscope enables sample magnification by up to 1000x, using visible wavelengths of...
Read More -

PMS Requirements of the EU MDR
on 31 January 2020 | By Stephen Biernacki
Our February 2020 webinar will help you optimize every element of PMS/PMCF as it relates to the EU MDR at your organization.
Read More -

Brexit: Top Five Do's and Don'ts for Medical Device Manufacturers
on 31 January 2020 | By Deven Snyder
After four years, many heated debates, several delays, and a few Prime Ministers later, the United Kingdom (U.K.) is officially leaving the European Union (EU) today, January 31, 2020. Well, sort of. Instead of the “hard Brexit” that had been promised and feared in March, April,...
Read More -

Med Device Monday: The iStent inject Device for Glaucoma Patients
on 27 January 2020 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

Developing an FDA Regulatory Strategy
on 8 January 2020 | By Stephen Biernacki
Our January 2020 webinar is designed to help you create a comprehensive regulatory strategy that enables you to successfully enter the US market.
Read More -

510(k): What is a Premarket Notification?
on 7 January 2020 | By Taryn Meade, Jordi Labs/RQM+ Director of Biological Evaluation
The Federal Food, Drug, and Cosmetics Act establishes strict requirements for devices and products marketed to consumers and professionals in the U.S. This includes medical devices, food and pharmaceutical packaging, and cosmetics containers. Before a new product can be...
Read More -

MDR’s May 2020 Deadline Won’t Change
on 6 January 2020 | By Deven Snyder
During a December meeting of the European Union’s Employment, Social Policy, Health and Consumer Affairs Council (EPSCO), European Commissioner for Health and Food Safety, Stella Kyriakides, confirmed that the May 2020 deadline for Medical Device Regulations (MDR) implementation...
Read More -

House Passes Bill to Repeal the Medical Device Tax
on 18 December 2019 | By Deven Snyder
The holidays may have just gotten a little happier and a lot brighter for those of us in the medical device industry! Earlier this week, the U.S. House of Representatives passed legislation to repeal the impending and much-criticized Medical Device Tax, a 2.3% excise tax on...
Read More -

Med Device Monday – FDA grants a new device that can detect HIV-1 Drug Resistance Mutations
on 9 December 2019 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

BREAKING NEWS: EU MDR Delay Class I Devices
on 27 November 2019 | By Nancy Morrison, RQM+
RQM+ clients have one more thing to be thankful for this year as a new Corrigenda to the European Union Medical Device Regulation (MDR) has been issued that extends the due date for Certificates for Class I – reusable surgical instruments – for four years. Check out the newly...
Read More -
R&Q Cares: Supporting Light of Life Rescue Mission
on 26 November 2019 | By Deven Snyder
The R&Q offices are buzzing with excitement as the holidays quickly approach! Many of us are preparing to gather with family and friends this week to celebrate Thanksgiving over a warm meal, football on the TV and great conversation with loved ones. As much as we are a company...
Read More -

Announcing R&Q's new Vice President of Technical Services, Ralph Asencio
on 25 November 2019 | By Stephen Biernacki
Regulatory & Quality Solutions (R&Q) is absolutely delighted to announce Ralph Asencio as Vice President of Technical Services! R&Q has plenty to be thankful for in 2019. It's been an incredible year for a multitude of reasons, one of which has been the addition of key technical...
Read More -

EU MDR/IVDR Guidance List Provides Insight on 2019 Expectations
on 7 November 2019 | By Deven Snyder
Get ready for an active fourth quarter in the European Medical Device Regulation (MDR) world! We sat down with R&Q's Nancy Morrison, Executive Director of Regulatory and Quality Consulting Services, to get her thoughts on the newly released "Ongoing Guidance development within...
Read More -

Q&A: FDA Updates for Medical Device Manufacturers
on 5 November 2019 | By Deven Snyder
From new software and cybersecurity guidance to updates to existing medical software policies, September was a busy month for FDA guidance releases. During our October webinar on FDA Updates for Medical Device Manufacturers, R&Q experts addressed these changes and what impacts...
Read More -
Med Device Monday: TransMedics OCS™ Lung System for Lung Preservation
on 28 October 2019 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

FDA Friday - A Closer Look at the CDRH Health of Women Program
on 18 October 2019 | By Erin Gontang
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

"Hope isn't a strategy." Takeaways from RAPS and new industry updates.
on 4 October 2019 | By Nancy Morrison, RQM+
September was an incredible month for us at R&Q. Our thought leaders were out and about at DeviceTalks Minnesota, our own 3-hour CER Virtual Workshop (now available on demand), The MedTech Conference (AdvaMed), and the RAPS Regulatory Convergence. Whew! If you participated in...
Read More -
Med Device Monday - XVIVO Perfusion System for Storing Human Organs Before Transplantation
on 30 September 2019 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

Solving the EU MDR Labeling Puzzle
on 17 September 2019 | By Stephen Biernacki
Our December webinar will help you understand and act on requirements.
Read More -

Strategies for Successful IVDR Implementation
on 17 September 2019 | By Stephen Biernacki
Our November 2019 webinar looked at how to assess products and more.
Read More -

FDA Updates
on 17 September 2019 | By Stephen Biernacki
Our October 2019 webinar highlighted key areas of change related to the FDA.
Read More -
What GMP Means in Pharma
on 11 September 2019 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
The acronym GMP refers to the good manufacturing practices promoted by the U.S. Food and Drug Administration (FDA) under the remit of the FD&C Act; which mandates strict levels of quality assurance at every stage of the pharmaceutical manufacturing chain. Although GMP principles...
Read More -

-
How to Conduct Molecular Weight Analysis
on 20 August 2019 | By Kevin Rowland, RQM+ Director of R&D
The molecular weight of proteins, natural and synthetic polymers is a clear indicator of product performance and activity. To control product performance across a broad range of applications, the understanding of molecular weight determination is key.
Read More -

Q&A: Integrating CERs and Post-Market Surveillance
on 27 June 2019 | By Stephen Biernacki
While we're in the midst of taking the summer off from webinars (see you in the fall!) and excitedly preparing for our CER Virtual Workshop in September, we did have our most popular and best-reviewed webinar ever in May: Integrating CERs and Post-Market Surveillance. The...
Read More -

What is Polymer Molecular Weight?
on 21 June 2019 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
Polymer molecular weight (MW) is the molecular mass of a polymer chain. While small molecules of the same elemental composition all exhibit the same molecular weight, polymer chains exhibit different characteristics. These chains are compounded from multiple small molecules that...
Read More -

Two sides of the regulatory coin
on 18 June 2019 | By Nancy Morrison, RQM+
One of the most rewarding aspects of regulatory work is that no two days are the same. There are always new (and constantly evolving) technologies and regulations to provide fresh opportunities to show your skills. If you’ve been tied up trying to get ready for May 26, 2020 and...
Read More -

Wednesday Wisdom: Article on Regulating Software as a Medical Device (SaMD)
on 5 June 2019 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

What is Elemental Analysis Used For?
on 22 May 2019 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
Elemental analysis is a process in analytical chemistry in which a sample of material such as water, minerals and bodily fluid is analyzed to ascertain its elemental composition. Elemental analysis can be qualitative, determining which elements are present or quantitative,...
Read More -

That's a wrap! A recap of our Advanced EU MDR and CER Workshops and what to do if you missed them.
on 9 April 2019 | By Stephen Biernacki
In March R&Q partnered with the Medical Alley Association in Minneapolis and MassMEDIC in Boston to offer industry-leading educational workshops on the EU MDR and CERs. R&Q recruited top industry experts, including representation from notified bodies BSI and GMED North America,...
Read More -

R&Q adds former TÜV Rheinland North America Lead Auditor of Medical to team
on 8 April 2019 | By Stephen Biernacki
Regulatory & Quality Solutions (R&Q) continues to grow aggressively – all while never sacrificing the high quality of service our clients have come to expect (this is a guiding principle and will never change) – and our latest addition to the team is particularly exciting. R&Q...
Read More -

EU MDR for Combination Products
on 25 March 2019 | By Stephen Biernacki
More companies will require notified body involvement.
Read More -

How to Choose a GPC Column
on 27 February 2019 | By Taryn Meade, RQM+ Director of Biological Evaluation Consulting
Gel Permeation Chromatography (GPC) is a powerful technique that provides a direct measurement of molecular weight, a critical property in characterizing natural and synthetic polymers. There is no universal GPC system with the capability to separate every polymer-based...
Read More -

Top 10 EU MDR and CER Questions
on 25 February 2019 | By Stephen Biernacki
Answers to the most popular questions we receive.
Read More -

EU MDR / CER Portfolio Planning
on 25 February 2019 | By Stephen Biernacki
Know the essential EU MDR portfolio planning requirements.
Read More -

QMS for EU MDR
on 25 February 2019 | By Stephen Biernacki
Does your quality system meet the additional requirements?
Read More -

Risk Management Updates
on 25 February 2019 | By Stephen Biernacki
What to do with your process to meet the EU MDR/IVDR requirements.
Read More -

Analyzing Solar Cells via X-ray Diffraction
on 1 February 2019 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
X-ray diffraction is a powerful method of characterizing crystalline materials in a non-destructive manner. It is often used to identify and quantify crystalline phases by illuminating samples with a beam of X-rays and analyzing the distinctive diffraction patterns emitted from...
Read More -
![🏅 Top 10 Questions: EU MDR and CER [Upcoming Webinar]](https://www.rqmplus.com/hs-fs/hubfs/RQ_Webinar_Promo_Top_10_1_21_19.png?length=450&name=RQ_Webinar_Promo_Top_10_1_21_19.png)
🏅 Top 10 Questions: EU MDR and CER [Upcoming Webinar]
on 25 January 2019 | By Stephen Biernacki
Are you registered for our next free webinar yet? It's one you won't want to miss and it's next week!
Read More -
Protein Characterization, Identification & Purification
on 25 January 2019 | By Kevin Rowland, Jordi Labs/RQM+ Director of R&D
Protein characterization is an incredibly broad field of study that encompasses a wide range of analytical methods and techniques. Although biochemists have made enormous strides in protein analysis, the process of identifying and purifying novel proteins remains a daunting...
Read More -

What does Brexit mean to your medical device company?
on 16 January 2019 | By Nancy Morrison, RQM+
Hope for the best, but prepare for the worst.
Read More -
Med Device Monday: The Fluobeam 800 and the PTeye System for Parathyroid Tissue Detection
on 7 January 2019 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

The Working Principles of Gel Permeation Chromatography
on 19 December 2018 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
Gel permeation chromatography (GPC) is a powerful analytical technique used to separate dissolved molecules by size, based on their elution from a column filled with a porous gel. I t can be described as a type of molecular sieving chromatography, where samples are separated...
Read More -
FDA Friday: Reclassification
on 30 November 2018 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

Premarket Notification: 510(k) FDA
on 21 November 2018 | By Taryn Meade, Jordi Labs/RQM+ Director of Biological Evaluation
A 510(k) is a premarket submission made to FDA to demonstrate that the device to be marketed is at least as safe and effective, that is, substantially equivalent, to a legally marketed medical device (21 CFR 807.92(a)(3)) that is not subject to PMA.1
Read More -

Our Advanced EU MDR and CER Workshop is coming to California in December at DeviceTalks West
on 14 November 2018 | By Stephen Biernacki
Note: Presenters/Panelists listed below subject to change. R&Q is bringing our Advanced EU MDR and CER Workshop to DeviceTalks West! Join us Dec.12 for an extensive, detailed look at EU MDR and CERs through the lens of top industry experts who have successfully implemented...
Read More -

Med Device Monday: Aethlon's Hemopurifier
on 29 October 2018 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

Polymer Testing: Polyethylene and End of Life Management
on 26 October 2018 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
Polyethylene is the most pervasive synthetic polymer used worldwide, comprising over a third of the world’s annual plastic manufacturing output. Production of plastics has come under significant environmental scrutiny in recent years with estimates suggesting that as much as...
Read More -

Using Transmission Electron Microscopy to Analyze Engineering Materials
on 10 October 2018 | By Kevin Rowland, RQM+ Director of R&D
Transmission electron microscopes (TEMs) are the big guns of the microscope world, capable of peering far beyond the diffraction limit of conventional optical microscopes to resolve sub-nanometer details and probe the atomic structure of materials. [1] The most sophisticated...
Read More -

What is Thermal Gravimetric Analysis?
on 8 October 2018 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
Thermogravimetric analysis, or thermal gravimetric analysis (TGA), is a method of determining the overall mass of a sample as a temperature-dependent property. Mass change as a function of temperature is a fundamental property of many materials as they degrade and lose volatile...
Read More -

FDA Friday: Updates to our Previous De Novo Pathway Blog
on 14 September 2018 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

New webinars, major events... hello, fall!
on 11 September 2018 | By Stephen Biernacki
First thing's first: our free webinars are back. Over the next three months – beginning September 25 – we'll be covering combination products, EU MDR, and economic operators. Slides, recordings, and Q&A will be made available to everyone who registers. We've made it easy for you...
Read More -

R&Q Q&A: Employee Spotlight and Career Opportunities
on 24 July 2018 | By Victoria Rose
This is our R&Q Q&A series. Every few months we ask R&Q employees how they make their impact at R&Q... and the impact R&Q has had on them. From improving people's lives to R&Q's rewarding culture, we hope their answers shed light on how our company influences the medical device...
Read More -

Not your average EU MDR and CER workshop
on 20 July 2018 | By Stephen Biernacki
Note: Presenters/Panelists listed below subject to change. DeviceTalks Boston is Oct. 8-10 this year and as part of it, R&Q is helping to organize an Advanced EU MDR and CER Workshop. It's an extensive, detailed look at EU MDR and CERs through the lens of top industry experts...
Read More -

Taking it up a notch at DeviceTalks Minnesota
on 10 May 2018 | By Stephen Biernacki
DeviceTalks Minnesota is right around the corner and we couldn't possibly be more excited - really. June 4-5 is only a little over three weeks away! We've witnessed the growth of this event first-hand over the past few years and this year we're confident will be the best yet,...
Read More -

🔑 Unlock the Secrets to CERs in our May Webinar
on 7 May 2018 | By Stephen Biernacki
Unlock the secrets to complying with the increased requirements for CERs in May's free R&Q webinar: CERs – Tips, Tricks, and Lessons Learned The session will be on Tuesday, May 22 from 1:00pm - 2:00pm EST. The presentation slides, webinar recording, and Q&A will be made...
Read More -
![💼 EU MDR – Proactive Post-Market Surveillance [Upcoming Webinar]](https://www.rqmplus.com/hs-fs/hubfs/Webinars_2018/April%202018/RQ_Webinar_EUMDR_Proactive_Post-Market_Surveillance_Promo-min%20%281%29.png?length=450&name=RQ_Webinar_EUMDR_Proactive_Post-Market_Surveillance_Promo-min%20%281%29.png)
💼 EU MDR – Proactive Post-Market Surveillance [Upcoming Webinar]
on 20 April 2018 | By Stephen Biernacki
It's time for our next free R&Q Intelligence Series webinar. The session – EU MDR – Proactive Post-Market Surveillance: The requirements and staff it will take to do it. – will be held Tuesday, April 24ᵗʰ from 1:00pm - 2:00pm EST. If you can't view the webinar live, we encourage...
Read More -

Typical Molecular Weights of Common Polymers
on 18 April 2018 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
No two polymers are the same. They exhibit different properties depending on key characteristics, such as their molecular weight. Polymer molecular weight (MW) describes the molar mass distribution of a polymer chain, which determines the weight of the macromolecule itself. This...
Read More -
What is Transmission Electron Microscopy?
on 13 April 2018 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
Transmission electron microscopy (TEM) is a form of microscopic analysis that transmits a high-energy beam of electrons through an ultrathin sample specimen. This provides imagery based on the transmission/attenuation of electrons, which is then magnified for direct observation...
Read More -

Wednesday Wisdom: Breakthrough Devices Program
on 11 April 2018 | By Blythe Sinclair
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -
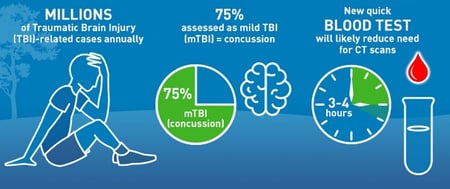
Med Device Monday: Diagnostic for Detection of Brain Trauma
on 2 April 2018 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

Polymer Failure Analysis with RQM+ Lab Services
on 30 March 2018 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
Failure analysis is used to detect chemical and mechanical defects and deficiencies in polymer products, which have contributed to or directly resulted in failure. It is a critical analytical field that can improve the efficiency and profitability of polymeric products,...
Read More -

R&Q Q&A: Employee Spotlight and Career Opportunities
on 6 March 2018 | By Victoria Rose
This is our R&Q Q&A series. Every few months we ask R&Q employees how they make their impact at R&Q... and the impact R&Q has had on them. From improving people's lives to R&Q's rewarding culture, we hope their answers shed light on how R&Q influences the medical device industry...
Read More -
Med Device Monday: Implantable Continuous Pump for Remodulin
on 6 March 2018 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

A Beginner’s Guide to Dynamic Mechanical Analysis
on 14 February 2018 | By By Kevin Rowland, RQM+ Director of R&D
Dynamic Mechanical Analysis (DMA) describes a procedure in which the mechanical properties of materials are measured as a function of time, temperature, and frequency. In basic terms, dynamic mechanical analysis involves the application of a stressor on a target such as the...
Read More -
Food Toxicology: Testing Food Contact Materials
on 1 February 2018 | By Kevin Rowland, RQM+ Director of R&D
A ubiquity of food contact materials exists today, with virtually all the food we consume coming packaged in some form or another. This has the multifaceted benefit of preserving food goods: maintaining quality for longer transit and storage periods; reducing the risk of...
Read More -

Measuring Polymer Exemptions with Gel Permeation Chromatography
on 15 January 2018 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
The US Environmental Protection Agency (EPA) was established to protect human health by providing regulatory oversight and implementing decisive action where required. Changes in manufacturing protocol and emergent materials mean that the powers of the EPA are routinely subject...
Read More -

FDA Friday - Michael Nilo, MS
on 12 January 2018 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

Webinar Q&A: Preparing your CER for MDR
on 8 January 2018 | By Stephen Biernacki
Note: R&Q's first Intelligence Series webinar of 2018 will be held on January 23rd – QMS for EU MDR: Does your quality system meet the additional requirements? Sign up here! -- Back in November and as part of the R&Q Intelligence Series, we held the webinar, Preparing your CER...
Read More -

Good Manufacturing Practice in the Medical Device Industry
on 5 January 2018 | By Kevin Rowland, RQM+ Director of R&D
All manufacturers are expected to establish some form of quality protocols to ensure that products meet a minimum standard, whether that threshold is established by general expectations or by regulators. Naturally, manufacturers of medical devices are held to a higher benchmark...
Read More -

Overcoming Uncertainty in E&L Testing
on 27 November 2017 | By Kevin Rowland, RQM+ Director of R&D
E&L testing is carried out on plastic products to understand which molecules, such as antioxidants, slip agents, surfactants lubricants, etc. are released from a polymer system. Undertaking E&L testing is a critical step of the medical device and pharmaceutical safety...
Read More -

Welcome, Jacob Foster: Regulatory and Quality Solutions' New Senior Principal Engineer
on 14 November 2017 | By Stephen Biernacki
Jacob Foster brings 20+ years of medical device and pharmaceutical regulatory, quality, and project management experience to R&Q. He also brings an engaging personality, eager to do more for clients. Pittsburgh, PA -- Regulatory and Quality Solutions LLC (R&Q) exists to improve...
Read More -
![Preparing your CER for MDR [Upcoming Webinar]](https://www.rqmplus.com/hs-fs/hubfs/Webinars/RQ/R%26Q%20Intelligence%20Series%202017/2017%20November%20-%20Preparing%20your%20CER%20for%20MDR/RQ_Webinar_Preparing_CER_for_MDR_Website_Promo_10_31_17-min.png?length=450&name=RQ_Webinar_Preparing_CER_for_MDR_Website_Promo_10_31_17-min.png)
Preparing your CER for MDR [Upcoming Webinar]
on 13 November 2017 | By Stephen Biernacki
The upcoming EU MDR requires enhanced clinical evidence to support the device whether you have Class I or Class III products. For products that have been on the market for a long time there may be limited or no clinical studies, and yet, the new European Medical Device...
Read More -

ISO 13485:2016 Companion Handbook Now Available
on 24 October 2017 | By Stephen Biernacki
ISO has published a companion handbook to ISO 13485:2016, Medical devices-Quality management systems - Requirements for regulatory purposes. From AAMI: It provides users with practical guidance and accurate interpretation of the requirements specified in the standard. It offers...
Read More -

Polyethylene (PE) High Temp GPC Separation Example
on 18 October 2017 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
New generation Resolve Columns (7.8 mm x 300 mm) are produced to allow separation over the molecular weight range from 500 to 12,000,000 g/mol (PS equivalent) for 13 µm particle size packing materials. Polyolefins often have broad molecular weight ranges characterized by high...
Read More -

Webinar Q&A: EU MDR / CER Portfolio Planning
on 2 October 2017 | By Stephen Biernacki
In September and as part of the R&Q Intelligence Series, we conducted the webinar, EU MDR / CER Portfolio Planning: Know the essential EU MDR portfolio planning requirements. Portfolio planning is a key aspect of interfacing with your notified body. The upcoming EU MDR changes...
Read More -

Webinar Q&A: Risk-Based Approach to ISO 13485:2016
on 19 September 2017 | By Stephen Biernacki
Last month and as part of the R&Q Intelligence Series we conducted the webinar, Risk-Based Approach to ISO 13485:2016: Risk considerations and implementation. Risk considerations and implementation of a risk-based approach in your QMS processes is one of the most significant...
Read More -
![EU MDR / CER Portfolio Planning: The Essential Requirements [Free Webinar]](//www.rqmplus.com/hs-fs/hubfs/Webinars/RQ/R&Q%20Intelligence%20Series%202017/September%20-%20Portfolio%20Planning/Webinar_Promo_September_Portfolio_Planning_Website-min.png?length=450&name=Webinar_Promo_September_Portfolio_Planning_Website-min.png)
EU MDR / CER Portfolio Planning: The Essential Requirements [Free Webinar]
on 12 September 2017 | By Stephen Biernacki
What? Please join us for September's free R&Q Intelligence Series webinar: EU MDR / CER Portfolio Planning: Know the essential EU MDR portfolio planning requirements. The session will take place Tuesday, September 26 from 1:00pm - 2:00pm EST. Portfolio planning is a key aspect...
Read More -

Pharmaceutical Lab Testing: APIs, Excipients, and Contact Materials
on 25 August 2017 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
Pharmaceuticals are designed, developed, and primarily manufactured in laboratory environments. Lab testing is a mandatory requirement in practically every phase of pharmaceutical R&D and production, from incoming raw materials inspection to dosage form quality control (QC)....
Read More -

MEDDEV 2.7/1 & CERs: Questions and Answers
on 21 August 2017 | By Stephen Biernacki
Clinical evaluation requirements have been changing, with the latest impact coming from MEDDEV 2.7/1 Rev 4. Preparing for and meeting these requirements is important because the grace period offered by some notified bodies is ending and clinical evaluation reports (CERs) are...
Read More -

Cybersecurity for Medical Devices: Questions and Answers
on 8 August 2017 | By Marilyn Waxberg
As the number of network connected medical devices increases, opportunities for improved patient care also increase. As a consequence, the vulnerability to cyber-attacks becomes a greater threat. Medical device cybersecurity threats can be dangerous for providers, networks, and...
Read More -

MDUFA IV: More Than Just User Fees - FDA Reauthorization Act (FDARA) Passes Senate
on 4 August 2017 | By Jake O'Donnell
Pending the expected signature of President Trump, MDUFA IV will be implemented (along with PDUFA VI and GDUFA II). Passage, including the associated rise in user fees for device manufacturers over the term of the act (FY18-22), was largely expected. Legislative politics involve...
Read More -

Clarifying the Clinical Evaluation Requirements: A Case Study
on 1 August 2017 | By Stephen Biernacki
About R&Q's Case Studies: We hope this Clinical Evaluation Report case study is valuable to you. Here are our currently available case studies. Subscribing to our blog is the best way to know when future case studies and other resources are available. Questions on this...
Read More -
Med Device Monday: Nonabsorbent Dressing
on 31 July 2017 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

EU MDR: Your Questions, Our Answers
on 27 July 2017 | By Stephen Biernacki
On May 5, 2017 the EU MDR (Medical Device Regulation) was published in the EU Official Journal. This date starts the three-year mandatory transition period where the MDR will replace the MDD (Medical Device Directive). The expanded scope of the MDR combined with the time frames...
Read More -

Regulatory and Quality Solutions Names Julie Maes Director of Territory Operations – Northern Lakes Region
on 24 July 2017 | By Stephen Biernacki
Maes brings 25+ years of medical device regulatory, quality, and project management experience to R&Q. Pittsburgh, PA -- Regulatory and Quality Solutions LLC (R&Q), a provider of industry-leading regulatory and quality engineering services to medical device and combination...
Read More -
Med Device Monday: Class II Device Types Exempt from Premarket Notification
on 18 July 2017 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -
E&L: Key Principles of Extractables & Leachables Analysis
on 14 July 2017 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
E&L testing is a critical process in pharmaceutical packaging development and medical device manufacturing. Unlike many other forms of laboratory analyses, it cannot be broken down into a series of simple methods. This is primarily due to the complex and infinitely variable...
Read More -

The Lowdown on Cybersecurity for Medical Devices
on 14 July 2017 | By Jyothsna Nunna
Every one of us has probably either been affected by or knows someone who has been affected by a cybersecurity vulnerability. Some attacks happen for the thrill, some to expose weaknesses between competitors, some for malicious intent, and some for money. We hear of these issues...
Read More -
![[Free R&Q Webinar] Cybersecurity for Medical Devices: The regulatory and quality ramifications.](//www.rqmplus.com/hs-fs/hubfs/Webinars/RQ/R&Q%20Intelligence%20Series%202017/July%20-%20Cybersecurity/Webinar_Promo_July_Cybersecurity_v5_Marilyn_Website-reduced.png?length=450&name=Webinar_Promo_July_Cybersecurity_v5_Marilyn_Website-reduced.png)
[Free R&Q Webinar] Cybersecurity for Medical Devices: The regulatory and quality ramifications.
on 6 July 2017 | By Stephen Biernacki
What? Please join us for a free R&Q Intelligence Series webinar: Cybersecurity for Medical Devices: The regulatory and quality ramifications. The session will be held on Tuesday, July 25 from 1:00pm - 2:00pm EST.
Read More -

Medical Device Monday: Tongue Depressors
on 4 July 2017 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

Med Device Monday: Bone Mills
on 30 May 2017 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -
What's Changed with FDA User Fees?
on 16 May 2017 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

RESOURCE: The MDR and IVDR Have Been Published! See Our Table of Key Dates
on 8 May 2017 | By Nancy Morrison, RQM+
Publication in the Official Journal of the European Journal on May 5, 2017 means the regulations will enter into force on May 26, 2017, twenty days after publication. We will have a three-year transition period for the MDR and a five-year transition period for the IVDR. Products...
Read More -
![MEDDEV 2.7/1 & CERs: Know the Changes and What to Do [Webinar]](//www.rqmplus.com/hs-fs/hubfs/Webinars/RQ/R&Q%20Intelligence%20Series%202017/May%20-%20MEDDEV/Webinar_Promo_May_MEDDEV_v2-min.png?length=450&name=Webinar_Promo_May_MEDDEV_v2-min.png)
MEDDEV 2.7/1 & CERs: Know the Changes and What to Do [Webinar]
on 2 May 2017 | By Stephen Biernacki
Part of your month, [almost*] every month: R&Q Intelligence Series webinars are held on the fourth Tuesday of every month. These free webinars from some of the medical device industry's leading-most experts are designed for reporting on and analyzing the top industry trends and...
Read More -

Titrimetry in Wine Analysis
on 19 April 2017 | By Kevin Rowland, RQM+ Director of R&D
Titrimetry is an important analytical process used to determine the quantity of specific constituents in a mobile phase. It is a type of volumetric analysis where the analyte is quantified as a function of equivalence, which is expressed as a titration curve with an end point...
Read More -

EU MDR and Clinical Evidence: What You Need to Know
on 18 April 2017 | By Ruthanne Vendy
THE SKINNY: The Council of the European Union adopted the proposed new European Medical Device Regulation (EU MDR) in March 2017 and passed by Parliament in April 2017. It is expected to be published in the Official Journal of the European Union in the upcoming weeks, making the...
Read More -
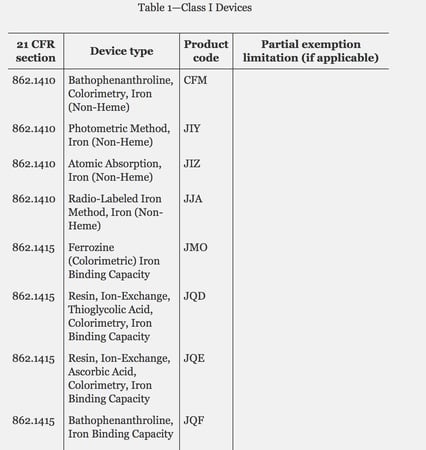
Med Device Monday: Class I Device Types Exempt from Premarket Notification
on 17 April 2017 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

Particulates and Residues in Medical Devices
on 12 April 2017 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
RQM+ Lab Services is one of the world’s leading authorities in the field of medical device analytical chemistry and laboratory testing. It is a deceptively broad field of analysis, comprising routine physical testing and complex product deformulation, alongside contamination and...
Read More -
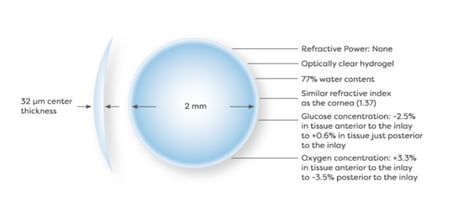
Med Device Monday: Raindrop Vision Inlay
on 11 April 2017 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

E&L Testing: Methods of Chemical Characterization
on 10 April 2017 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
Extractables and leachables testing – E&L testing for short – is the process of identifying and quantifying the molecules which can be released from plastic-containing products. This blog post discusses the many different analytical techniques which are used to analyze sample...
Read More -
![EU MDR: Assessing the Impact and Next Steps [Webinar]](//www.rqmplus.com/hs-fs/hubfs/Webinars/RQ/R&Q%20Intelligence%20Series%202017/April%20-%20EU%20MDR/Webinar_Promo_April_EU_MDR_v2-min.png?length=450&name=Webinar_Promo_April_EU_MDR_v2-min.png)
EU MDR: Assessing the Impact and Next Steps [Webinar]
on 5 April 2017 | By Stephen Biernacki
Part of your month, every month: R&Q Intelligence Series webinars are held on the fourth Tuesday of every month. These free webinars from some of the medical device industry's leading-most experts are designed for reporting on and analyzing the top industry trends and topics in...
Read More -

Exploring the Importance of Polymer Analysis
on 4 April 2017 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
Polymers are ubiquitous materials used in construction, engineering, and countless commercial products. These applications can be as varied as the polymeric materials used in medical devices, pharmaceuticals, food and beverage packaging, and personal care cosmetics. They are...
Read More -
Contamination Detection: Particulates and Residue Analysis with RQM+ Lab Services
on 31 March 2017 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
Process and quality control in the pharmaceutical industry requires highly-discerning contaminant detection, including particulates and residue analysis. These elements are among the primary forms of contamination in the pharmacological industry, and can arise from a broad range...
Read More -

What is ISO 10993 Used for?
on 29 March 2017 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
ISO 10993 comprises a series of international standards for the evaluation of biomedical devices and associated biological risk. ISO 10993 includes specific standards for certain material classes, such as ceramics or metals, as well as evaluation and testing within a...
Read More -

Med Device Monday: C-Section Retractor
on 28 March 2017 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

Carrying Out Particle Analysis Using DLS
on 20 March 2017 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
There are various well-established methods of carrying out particle analysis including dynamic image analysis (DIS), static light scattering (SLS), and sieve analysis—sometimes called gradation testing. These methodologies can often be complementary, each providing information...
Read More -

MDSAP: A Better Inspection Option for Device Manufacturers?
on 14 March 2017 | By Jake O'Donnell
Note: This post is written by Jake O'Donnell, Senior FDA Compliance Principal at R&Q. Jake has more than 20 years of experience working for the FDA. Please join us for a free one-hour webinar on MDSAP, March 28th. Medical device firms, are you looking forward to your next FDA...
Read More -
![MDSAP: What is it? Is it for me? How will I get it done? [Webinar]](//www.rqmplus.com/hs-fs/hubfs/Webinars/RQ/R&Q%20Intelligence%20Series%202017/March%20-%20MDSAP/Webinar_Promo_March-min.png?length=450&name=Webinar_Promo_March-min.png)
MDSAP: What is it? Is it for me? How will I get it done? [Webinar]
on 7 March 2017 | By Stephen Biernacki
Part of your month, every month: R&Q Intelligence Series webinars are held on the fourth Tuesday of every month. These free webinars from some of the medical device industry's leading-most experts are designed for reporting on and analyzing the top industry trends and topics in...
Read More -

Review Controls Applied to Outgoing Data Streams (Because the FDA Might Be)
on 6 March 2017 | By Jake O'Donnell
Medical device manufacturers need to carefully manage several important outward streams of information regarding their products. The regulatory community is generally most familiar with the data stream that relates to obtaining clearance to market devices, which involves...
Read More -

NEWS: Bill Introduced to Improve FDA's Medical Device Inspection Process
on 1 March 2017 | By Jake O'Donnell
Senator Johnny Isakson [R-GA] Note: This post is written by Jake O'Donnell, Senior FDA Compliance Principal at R&Q. Jake has more than 20 years of experience working for the FDA. We hope his commentary brings a useful perspective to the new bill. Anytime there is an...
Read More -
UV-VIS Absorbance and Protein Characteristics
on 16 February 2017 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
One of the most common methods for analyzing protein characteristics and measuring protein purity in solution is to observe the sample’s absorption of ultraviolet (UV) wavelengths of light. Proteins that contain the appropriate amino acids are absorbent to light on the...
Read More -
![A Risk-Based Approach to Your QMS Implementation - ISO 13485:2016 and ISO 9001:2015 [Webinar]](//www.rqmplus.com/hs-fs/hubfs/Headshots/Optimized_for_Web/mark_swanson-min.jpg?length=450&name=mark_swanson-min.jpg)
A Risk-Based Approach to Your QMS Implementation - ISO 13485:2016 and ISO 9001:2015 [Webinar]
on 12 February 2017 | By Stephen Biernacki
Part of your month, every month: R&Q Intelligence Series webinars are held on the fourth Tuesday of every month. These free webinars from some of the medical device industry's leading-most experts are designed for reporting on and analyzing the top industry trends and topics in...
Read More -

An Overview of Toxicological Risk Assessments
on 25 January 2017 | By Taryn Meade, RQM+ Director of Biological Evaluation Consulting
What is a Toxicological Risk? Toxicological risk measures the probability of an adverse effect being caused by a given compound. This covers effects such as skin irritation from topical treatments to more serious risks such as a material being carcinogenetic or fatal if ingested.
Read More -

Acquisition Regulatory Assessments: A Case Study
on 25 January 2017 | By Stephen Biernacki
About R&Q's Case Studies: We hope this Acquisition Regulatory Assessments case study is valuable to you. Here are our currently available case studies. Subscribing to our blog is the best way to know when future case studies and other resources are available. Questions on this...
Read More -

Med Device Monday: Restless Leg Relaxer
on 24 January 2017 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

R&Q Q&A: Minnesota Spotlight
on 20 January 2017 | By Victoria Rose
Welcome to the third post in our R&Q Q&A series. Every few months we ask R&Q employees from a different region questions about how they make their impact at R&Q... and the impact R&Q has had on them. From improving people's lives to R&Q's corporate culture, we hope their answers...
Read More -
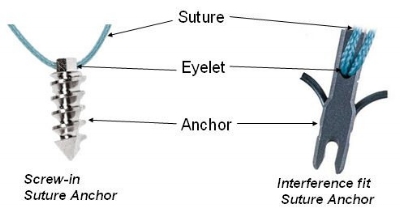
Med Device Monday: Bone Anchors
on 10 January 2017 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

Regulatory and Quality Solutions (R&Q) Certified By the Women’s Business Enterprise National Council
on 9 January 2017 | By Stephen Biernacki
R&Q, provider of industry-leading regulatory and quality consulting and engineering services to medical device companies, is proud to announce national certification as a Women’s Business Enterprise by the Women's Business Enterprise Council-PA-DE-sNJ, a regional certifying...
Read More -

A Cool New Way to Prevent Cancer Hair Loss — Literally
on 19 December 2016 | By Kate Keverline
♫ Baby, It's Cold Outside, especially in Texas, where a clinical trial showed a new cooling cap system can prevent hair loss in cancer patients. The ongoing trial involved 95 women with early stage breast cancer. After four cyles of chemotherapy, 48 of the patients—or 51...
Read More -

Headway in PA: Announcing Dedicated Operations Directors in Pittsburgh and Philadelphia
on 16 December 2016 | By Stephen Biernacki
In 2016 R&Q expanded its activities and the amount of clients served in the northeast (specifically, the greater Boston region), formally launched R&Q in Minnesota, and continued to service new and existing clients throughout the state of Ohio. In 2017 we plan to strengthen our...
Read More -

Process Validation Case Study: Know What You Know... and What You Don't
on 13 December 2016 | By Stephen Biernacki
About R&Q case studies: We hope this process validation case study helps you understand how we serve our medical device clients in this area. If you have specific process validation questions please contact us and we'll be happy to help. Looking to solve a different problem?...
Read More -

How to Develop a Biological Evaluation Plan
on 17 November 2016 | By Taryn Meade, RQM+ Director of Biological Evaluation Consulting
A biological evaluation plan is developed to systematically assess whether a device intended for use in humans poses an unacceptable biological safety risk.
Read More -

Vital To-Do for 2017: Prepare for ISO 13485:2016
on 7 November 2016 | By Stephen Biernacki
“Procrastination is the bad habit of putting of until the day after tomorrow what should have been done the day before yesterday.” - Napolean Hill It's really never too early, especially when it comes to renewing the certifications you need to keep your product on the market. In...
Read More -

Slides and Webinar Recording: Chemical Characterization Requirements
on 28 October 2016 | By Stephen Biernacki
Missed our webinar with iuvo BioScience - Chemical Characterization Requirements: ISO 10993-18 and Extractables & Leachables Requirements? Good news: you can now download the slides and view the recording.
Read More -

Next Up: FDA Regulatory 101 with Combination Products Spotlight at Heal Ohio Conference
on 19 October 2016 | By Stephen Biernacki
This is the last FDA Regulatory 101 installment of the year for our partnership with BioOhio. After events in Cincinnati and Columbus, the October 27th event will be held during the 2016 Heal Ohio Conference at the University of Akron. Former FDA Consumer Safety Officer Jake...
Read More -

Engineering Plastics via Gel Permeation Chromatography
on 14 October 2016 | By Kevin Rowland, RQM+ Director of R&D
Gel permeation chromatography (GPC) is a well-known method which is used for characterizing polymers according to their molecular weight distribution. The gel permeation chromatography analysis of engineering plastics at high temperatures needs specialized instruments and...
Read More -

Master 510(k), IDE, and PMA Submissions at AdvaMed's Workshops
on 14 October 2016 | By Kate Keverline
Within driving distance to D.C.? There's still time to enjoy a few days there next week and simultaneously hone your submissions skills. The FDA, R&Q, and other talented industry experts courtesy of AdvaMed are offering three workshops dedicated to teaching you the guidelines...
Read More -

CAPA on CAPA: Webinar Slides and Recording
on 7 October 2016 | By Stephen Biernacki
Missed our CAPA on CAPA webinar with MassMEDIC? Now you can download the slides and view the recording.
Read More -

The REdI Fall 2016 Conference: Slides and Recordings
on 5 October 2016 | By Kate Keverline
The Regulatory Education for Industry (REdI) Conference is an FDA-led forum that brings together the regulatory educators from FDA’s Center for Drug Evaluation and Research (CDER) and Center for Devices and Radiological Health (CDRH) to provide direct, relevant, and helpful...
Read More -
Med Device Monday: Briteseed and SafeSnips
on 3 October 2016 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

Free Webinar: Chemical Characterization Requirements: ISO 10993-18 and Extractables & Leachables Requirements
on 28 September 2016 | By Stephen Biernacki
What? This joint webinar presented by Regulatory and Quality Solutions (R&Q) and iuvo BioScience is focused on the requirements for chemical characterization in the healthcare industry. Specifically, it will discuss the standards required for ISO 10993-18 chemical...
Read More -

R&Q Q&A: Northeast Employee Spotlight and Openings
on 23 September 2016 | By Victoria Rose
Welcome to the second post in our R&Q Q&A series. Every few months we ask R&Q employees from a different region questions about how they make their impact at R&Q... and the impact R&Q has had on them. From improving people's lives to R&Q's corporate culture, we hope their...
Read More -

Minnesota Momentum: R&Q Hires Member of ISO Technical Committee 210, Mark Swanson
on 21 September 2016 | By Stephen Biernacki
Mark Swanson is R&Q's new Director of Minneapolis–Saint Paul (MSP) Territory. Welcome, Mark! Mark's addition firmly establishes R&Q in the Twin Cities and prepares R&Q to handle growth in the region. The hiring also builds upon R&Q's existing technical leadership, increasing...
Read More -

Revamping Supplier Quality: A Case Study
on 12 September 2016 | By Stephen Biernacki
About R&Q's Case Studies: We hope this Supplier Quality Remediation case study is valuable to you. Here are our currently available case studies. Subscribing to our blog is the best way to know when future case studies are available. Questions on supplier quality remediation?...
Read More -

When CAPA Needs a Corrective Action: A CAPA Remediation Case Study
on 8 September 2016 | By Stephen Biernacki
About R&Q's Case Studies: We hope this case study is valuable to you. Here are our currently available case studies. Subscribing to our blog is the best way to know when future case studies are available. Questions? Contact us. Challenge The FDA found problems you didn't think...
Read More -

Medical Device Development: Avoiding Pitfalls in Human Factors / Usability and Design
on 7 September 2016 | By Carol Vierling
This article was originally published in Summer of 2016's Made in PA issue, a publication of the Pittsburgh Technology Council. The competitive landscape of medical devices combined with the advantages of being first to market often tempt manufacturers to move through the...
Read More -

Med Device and Med Tech Startups: Master Regulatory Basics and More! September 22 in Philly
on 1 September 2016 | By Stephen Biernacki
We all gotta start somewhere... and Pennsylvania Bio and Quorum have helped us put together an invaluable event to help your early-stage medical device company hit the ground running. Seasoned experts, such as R&Q's President Maria Fagan and Buchanan Ingersoll & Rooney's Will...
Read More -

5 Reasons Not to Fear a Supplier Audit
on 30 August 2016 | By Michelle Mahoney
The following post is written by R&Q Senior Engineer, Michelle Mahoney. One of the toughest things to experience when walking in to audit a company is the tension, anxiety, and fear emanating from those in the room. Taking a look around the room, I notice heads are turned away,...
Read More -
What You Should Do About the Two New FDA Draft Guidance Documents
on 24 August 2016 | By Nancy Morrison, RQM+
It has been a long journey for the FDA to get the new draft guidances published on when to submit a 510(k) for a change to an existing device, and another specific to software changes. But on August 8th, 2016 it finally happened! For those of you who remember the FDA's attempt...
Read More -

Free Webinar: CAPA on CAPA, Presented by MassMEDIC and R&Q - Register Now!
on 16 August 2016 | By Stephen Biernacki
CAPA is often overlooked as an integral component of a quality system. More often than not, the FDA can trace deficiencies in a system directly back to the CAPA system. However, an effective quality system needs more than just a rework of the outcome of the ineffective CAPA...
Read More -
Med Device Monday: Prosthetic Face
on 16 August 2016 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

R&Q's Christine Santagate Featured in Medical Design & Outsourcing
on 3 August 2016 | By Stephen Biernacki
Staying at the forefront of the medical device industry takes work - a lot of it, in fact. Communication with R&Q's peers in the industry is in large part how our employees do it, and a big part of that is addressing hot topics in the industry. R&Q's own Client Solutions Advisor...
Read More -
Preparing for New Regulations of Elemental Impurities in Drugs
on 1 August 2016 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
Drug and dietary supplement manufacturers face an increasingly complex set of elemental impurity standards, which we will detail here.
Read More -

July is a BIG month for R&Q in Columbus, OH
on 12 July 2016 | By Stephen Biernacki
■ ■ ■ Are you an Ohio Bioscience Expo & Showcase attendee here to register for the FDA Regulatory 101 Series? ■ ■ ■ The Ohio Bioscience Expo & Showcase and next FDA Regulatory 101 Series event are on July 27th and 28th, respectively. If you're outside of Columbus but in Ohio,...
Read More -

Wearables are Exploding (Not Literally!) and Transforming the Medical Device Industry
on 8 July 2016 | By Kate Keverline
Image via Verily Smart wearables are turning into a largely profitable industry. From glucose monitoring, to sleep technology, to fitness apps, the number of wearable medical devices on the market are continuing to grow and in turn, help the lives of people - with or without...
Read More -

10 Tips for Adopting the Updated ISO 13485:2016 Standards: Webinar Slides and Recording
on 7 July 2016 | By Stephen Biernacki
Missed our latest webinar on this year's ISO 13485 changes? Now you can download the slides and recording. R&Q's Director of Regulatory Affairs Nancy Morrison and Client Solutions Advisor Christine Santagate walked through 10 tips for adopting the latest changes. From conducting...
Read More -

What is the Karl Fischer Titration Method?
on 6 July 2016 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
At RQM+ Lab Services , our team of expert lab technicians utilize a range of testing methods to deliver precise results for specific customer requirements. Where water determination is the primary ob j ective, we often deploy the Karl Fischer titration method; the...
Read More -

FDA Approved: A Human Factors and Usability Case Study
on 22 June 2016 | By Stephen Biernacki
About R&Q's Case Studies: We hope you enjoy our latest case study. View all of our available Case Studies. Subscribing to our blog is the best way to know when future case studies are available. FDA Approved: Creating and Implementing a Compliant Usability Engineering Process...
Read More -

4 Things R&Q Learned At MD&M East
on 21 June 2016 | By Stephen Biernacki
What an action-packed three days MD&M East was this year! Our team is exhausted, but in the absolute best way possible. I'm not sure how anyone who attended the show could've escaped the exhaustion... as the exhibit floor was BIG. If you're reading this and attended, hopefullly...
Read More -

This New Med Device Will Help You Sleep Better
on 17 June 2016 | By Kate Keverline
Everyone appreciates a good night's sleep. A big obstacle to that is the fact that 18 million American adults suffer from Obstructive Sleep Apnea (OSA), and the condition arises simply from a wayward throat muscle. A new study by Dr. Richard Schwab, co-medical director of the...
Read More -

Industry Advice: Earn RAC Points and Enhance Your Resume by Writing for RAPS' Regulatory Focus
on 13 June 2016 | By Carol Vierling
You've already read a few benefits in the title but let's reiterate: Earn RAC points Enhance your resume/LinkedIn profile Get published, and share your expertise with industry peers in the process These are just three of the several reasons you should consider writing an article...
Read More -
The de novo Pathway: Is It Right for My Device?
on 10 June 2016 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

R&Q Q&A: Boston Employee Spotlight
on 8 June 2016 | By Kate Keverline
Welcome to the first post in our R&Q Q&A series. Every two months we'll ask R&Q employees from a different region questions about how they make their impact at R&Q... and the impact R&Q has had on them. From improving people's lives to R&Q's corporate culture, we hope their...
Read More -
Wellness Devices vs. Medical Devices
on 3 June 2016 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -
The Med Device Industry (and the FDA) Embrace 3D Printing Innovation
on 25 May 2016 | By Kate Keverline
Just this week the world's first 3D-printed office opened in Dubai. It took all of 17 days to print a 2,700-square-foot building at half the price of conventional methods. In short, this 3D printing thing has some legs... and we mean that literally, too. 3D printing is...
Read More -

R&Q: Engineered by Women
on 24 May 2016 | By Stephen Biernacki
Did you know that women are half as likely as men to start businesses? That's a statistic R&Q wants to help change. Aside from the importance of gender and fairness issues, women entrepreneurship fuels economic growth. R&Q is Engineered by Women. We're proud to have been started...
Read More -
A Whole New World of UDI: Slides and Recording
on 20 May 2016 | By Stephen Biernacki
If you have any UDI needs or questions, do not miss this post.
Read More -

MD&M East Expo and Conference: June 14-16
on 17 May 2016 | By Kate Keverline
Be a part of the largest and most influencial medical manufacturing event on the east coast. Join more than 500 leading medtech suppliers and manufacturers - including Johnson & Johnson, Stryker, and countless others - at the Medical Design & Manufacturing (MD&M) East Expo and...
Read More -

How I Broke into Regulatory Affairs, and How You Can, Too
on 13 May 2016 | By Allison Komiyama, Ph.D., RAC, RQM+ Vice President, MedTech Innovations
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of...
Read More -

Presentation and Networking at PA Bio: Practical Considerations for Avoiding FDA Compliance Problems (BEING RESCHEDULED)
on 11 May 2016 | By Stephen Biernacki
NOTE: This event has been cancelled and is being rescheduled. Organizer Regulatory and Quality Solutions (R&Q) What? A presentation and networking event over lunch at PA Bio: Practical Considerations for Avoiding FDA Compliance Problems, and How to Manage Them If They Arise In...
Read More -

The New ISO 13485 Standards are Here! Sign Up for a Free R&Q Webinar
on 11 May 2016 | By Stephen Biernacki
The world's most well known standards for medical device quality management have been updated for the first time since 2003. Released in March by the International Organization for Standardization, ISO 13485 provides the universal requirements for manufacturers and service...
Read More -

AdvaMed Workshop: Integrating Human Factors into Med Device Design Control and Beyond
on 4 May 2016 | By Kate Keverline
Attention any and all regulatory affairs specialists, engineering managers, quality & production engineers, QA/Q directors and managers and compliance officers: AdvaMed is presenting a Design Control-focused workshop in Golden Valley, Minnesota, June 7-8. The workshop has been...
Read More -
MassMEDIC Webinar: A Whole New World of UDI - May 18
on 28 April 2016 | By Stephen Biernacki
If you missed R&Q's Paul Robinson discuss UDI during R&Q's afternoon workshop at BIOMEDevice in April, here's a second chance. A similar presentation and discussion will be offered in conjunction with MassMEDIC on Wednesday, May 18th at 11:30am EST. All the details - including...
Read More -

PA Bio Connect@ - May 9 in Philadelphia
on 25 April 2016 | By Kate Keverline
Great news, Philly! EisnerAmper will be hosting the Pennsylvania Bio: Connect@ Series May 9th in Philadelphia - and we'll be there. R&Q is a longstanding member of Pennsylvania Bio and is excited to network at this event and other upcoming events in the area, too. Here's what to...
Read More -

AdvaMed Webinar: Avoiding and Managing FDA Compliance Problems - Monday, May 9th
on 15 April 2016 | By Stephen Biernacki
Did you miss former FDAer Jake O'Donnell speak about avoiding and managing FDA compliance problems during R&Q's afternoon workshop at BIOMEDevice earlier this week? If so, we have news especially for you: he will be presenting on a similar topic via a webinar on Monday, May 9th...
Read More -
7 Methods of Assessing Protein Purity
on 14 April 2016 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
Just one mammalian cell is made up of a combination of many distinct proteins, often to the degree of thousands in varying concentrations. The variety and magnitude of protein types within just micro volumes of a single sample micro present substantial challenges for...
Read More -

NEW White Paper - Practical Considerations for Avoiding Regulatory Escalation: R&Q's Latest, In-Depth White Paper from a Former FDA Consumer Safety Officer
on 13 April 2016 | By Stephen Biernacki
There’s a formula for effectively managing an adverse FDA inspection and the newest R&Q white paper shares it. Learn all the practical considerations for avoiding regulatory escalation from a former FDA Consumer Safety Officer: R&Q's Jake O'Donnell. This is your free, ultimate...
Read More -

Minnesota, Here We Come: See You at the 2016 Medical Alley Annual Meeting, April 27th
on 23 March 2016 | By Stephen Biernacki
R&Q will be a Supporting Sponsor at the Medical Alley Association's 2016 Annual Meeting, April 27th. If you're not familiar, the association is a state-based member organization servicing the health technology community who works to promote Minnesota's Medical Alley by...
Read More -

R&Q's Can't-Miss Education Event of the Year: An Afternoon Training Workshop at BIOMEDevice in Boston, April 13th
on 8 March 2016 | By Stephen Biernacki
Those in the northeast, get ready! Not only will R&Q be exhibiting at this year's BIOMEDevice show (booth 640) and sponsoring MassMEDIC's Annual Conference, but we'll be presenting an afternoon training workshop (three separate sessions) free to attendees of the BIOMEDevice show...
Read More -

When Benefit Outweighs Risk: Creating a Successful Clinical Evaluation Report - A Case Study
on 18 February 2016 | By Stephen Biernacki
About R&Q's Case Studies: We hope you enjoy our third case study. View all of our available Case Studies. Subscribing to our blog is the best way to know when future case studies are available. Challenge High volume of data... and differing opinions. A client's product is...
Read More -

5 Heads are Better Than 1: De-Risking Your Regulatory Pathway Using a Team Approach - A Case Study
on 15 February 2016 | By Stephen Biernacki
About R&Q's Case Studies: We hope you find value in our second case study. View all of our available Case Studies. Subscribing to our blog is the best way to know when future case studies are available. Challenge Regulation doesn't have to be a barrier to innovation. A client...
Read More -

Introducing Our First In A Series of New RQM+ Case Studies: Design History File Remediation
on 12 February 2016 | By Stephen Biernacki
About RQM+ Case Studies: Our goal in producing case studies is to succinctly demonstrate how RQM+ applied our unique expertise to tackle a client's challenge, implement a competent solution, and demonstrate real results. We will post each case study to our blog and offer a...
Read More -

Announcing the 2016 FDA Regulatory 101 Series
on 8 February 2016 | By Stephen Biernacki
Medical device professionals in Ohio, are you ready? Last year R&Q teamed with BioOhio to present an ongoing series of 101 medical device events throughout the year. Thanks to participation and feedback from ambitious medical device professionals across Ohio - and the support of...
Read More -

Hear R&Q and Other Industry Experts Discuss 510(k) Submissions at AdvaMed's Workshop In Irvine, CA
on 2 February 2016 | By Stephen Biernacki
Join R&Q's Marilyn Waxberg and Nancy Morrison as they speak along with other industry experts at AdvaMed's 510(k) Submissions Strategy Workshop in Irvine, CA this month. The event is scheduled for two days: February 22-23. Marilyn is R&Q's Senior Principal Advisor and Nancy is...
Read More -

Biological Endpoint Testing in Biocompatibility Evaluations
on 1 February 2016 | By Taryn Meade, RQM+ Director of Biological Evaluation Consulting
Biological safety evaluations are designed to assess whether a product is safe for its intended use depending on its composition. For example, an evaluation of a cardiovascular implantable device would need to assess potential risks associated with long term contact with blood...
Read More -
Revisiting a Classic: The Pirate-Themed CT Scanner
on 26 January 2016 | By Stephen Biernacki
Behold medical device ingenuity at its finest. In 2013 the NewYork-Presbyterian Morgan Stanley Children's Hospital purchased a pirate-themed CT scanner, and the result is an incredible way to put children at ease before being scanned. See the rest of the creativity in pictures...
Read More -

R&Q Is Sponsoring The M2D2 $100K Challenge and A Summary of Important Dates
on 22 January 2016 | By Stephen Biernacki
The University of Massachusetts Medical Device Development Center (M2D2) has announced the M2D2 $100K Challenge 2016, a nationwide competition that showcases innovative ideas of early-stage medical device, diagnostic, and biotech companies. R&Q is proud to announce that we are...
Read More -

Presentation and Networking: Is A Medical Device Hiding In Your Product Portfolio? Join R&Q at MassBio!
on 14 January 2016 | By Stephen Biernacki
Organizer Us! Regulatory and Quality Solutions (R&Q) What? A presentation and networking event over lunch addressing how an accessory or tool, in reality, is often a medical device. This session will discuss combination products and the use of accessories with drugs and...
Read More -

Announcing R&Q's Newest Senior FDA Compliance Principal: Former FDA Consumer Safety Officer Jake O'Donnell
on 6 January 2016 | By Stephen Biernacki
R&Q is thrilled to announce the newest member of our team. Jake O'Donnell will act as Senior FDA Compliance Principal beginning February 2016. Jake formerly acted as Consumer Safety Officer (CSO) at the FDA for 20 years.
Read More -

Chemical Characterization Testing of Medical Devices
on 4 January 2016 | By Taryn Meade, RQM+ Director of Biological Evaluation Consulting
The Food and Drug Administration (FDA) partially recognized the ISO 10993 guidance making it one of the most important for the chemical characterization testing of medical devices.1 ISO 10993 outlines all of the necessary steps for regulatory compliance to use new or existing...
Read More -
We Made a Video About R&Q! Here's Why.
on 1 December 2015 | By Stephen Biernacki
One of R&Q's priorities in 2015 was to find new, distinctive ways to tell our story. We wanted to make it abundantly clear to potential clients and employees that R&Q is first and foremost about improving lives. We also wanted to clearly explain how we improve lives. The result...
Read More -

Welcome To R&Q's New Website!
on 13 November 2015 | By Stephen Biernacki
Welcome to R&Q's updated website! To state it plainly, we've revamped our website to better serve the needs of regulatory and quality professionals in the medical device industry. We hope you find it easy to use. Some changes include more clearly outlined services, additional...
Read More -

Common Mistakes in Extractables and Leachables Analysis
on 2 November 2015 | By Kevin Rowland, RQM+ Director of R&D
An important step in determining the biological compatibility and safety of a medical device as required by ISO 10993 is chemical characterization. Extractables and leachables testing is a vital but complex part of this. There is no one-size-fits-all approach that can be...
Read More -

Determination of Protein Molecular Weight
on 7 August 2015 | By Kevin Rowland, RQM+ Director of R&D
Determining the structural, chemical and functional properties of proteins is of vital importance to an increasing range of biopharmaceutical and life science markets. This is conducted repeatedly throughout the research and development (R&D) process for initial characterization...
Read More -
Notified Body Expectations: Today, Tomorrow and into the Future
on 27 May 2015 | By Pauling Panagis
R&Q and Nerac invite you to attend a free medical device event and panel discussion titled: Notified Body Expectations: Today, Tomorrow and into the Future Space is Limited - Register Today https://www.regonline.com/builder/site/Default.aspx?EventID=1718288 Thursday, June 18,...
Read More -

Identifying Particulates and Residue in Pharmaceuticals
on 19 May 2015 | By Kevin Rowland, RQM+ Director of R&D
Pharmaceuticals are subject to the strictest controls – and rightly so. International and regional regulators work to mandate the highest quality standards for therapeutics and medical devices, which puts a real burden of responsibility on manufacturers and packagers.
Read More -
Christmas Tree: Artificial or Real
on 15 April 2015 | By Kevin Rowland, Jordi Labs/RQM+ Director of R&D
Making the choice between a real or ‘fake’ Christmas tree can be a tough family decision. While there are many reasons to choose artificial (no needles, doesn’t dry out, cheaper over time), possibly the best reason is safety.
Read More -
What Extractable and Leachable Compounds are Found in Syringes?
on 27 March 2015 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
Extractables and leachables are chemicals that can be extracted from a product such as a syringe and deposited into the product it is housing. This can be either in the presence of a solvent or through direct contact over an extended period of time. An extractable and leachable...
Read More -

Product Deformulation: Reverse Engineering with RQM+ Lab Services
on 11 March 2015 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
Product deformulation is a complex analytical process designed to uncover the specific chemical identity of finished goods. Chemists take the product of interest, break it down into its constituent parts, separate those into distinct entities, then analyze both the major and...
Read More -

Applications of Thermal Gravimetric Analysis
on 10 March 2015 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
Thermo gravimetric analysis (TGA) is a thermal analysis technique used to quantify the overall mass of a sample as a temperature dependent property. The change in mass as a function of temperature is a key property in a range of materials because they lose volatile components as...
Read More -
E&L For Bis(2,4-di-tert-butylphenyl)-phosphate (bDtBPP)
on 6 March 2015 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
Bis(2,4-di-tert-butylphenyl)-phosphate (bDtBPP) has been the subject of much discussion in the area of Extractables and Leachables testing for single use bioprocess systems.
Read More -

Product Deformulation: A Step-by-Step Guide to Reverse Engineering
on 10 December 2014 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
Chemical reverse engineering is an extremely valuable process for those looking to protect their intellectual property from manufacturing issues or patent infringements. It is also deployed to help manufacturers maintain a competitive edge through targeted product analysis. Each...
Read More -
Common Causes of Polymer Failure
on 1 December 2014 | By Kevin Rowland, RQM+ Director of R&D
Understanding how and why materials degrade and ultimately fail is a critical step in the research and development (R&D) process. Compared to the lifespan of traditional engineering materials, polymers tend to fail in comparatively short timeframes. It is one of the reasons that...
Read More -

Contaminant Analysis: How to Interpret Particulate Matter
on 7 October 2014 | By Kevin Rowland, RQM+ Director of R&D
Particulate contamination analysis using microscopy and spectroscopy methods can identify and mitigate contamination in chemical, consumer products, industry or processes. Particulate analysis is the first stage in resolving contamination issues that could be potentially...
Read More -

New Trends in Analytical Chemistry – The Corona Charged Aerosol Detector (CAD)
on 17 September 2014 | By Kevin Rowland, RQM+ Director of R&D
The world of liquid chromatography has come a long way since the first online detectors for HPLC systems became available. At that time sixty some years ago, refractive index and conductivity detectors were all that was available, but neither provided the sensitivity that would...
Read More -
Molecular Weight Determination of Pharmaceuticals with Novel GPC
on 27 August 2014 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
Molecular weight is a key property used in the characterization of polymer-based products. It refers to the molar mass distribution of polymer chains which can vary widely within polymers of the same chemistry. This can be problematic for regulated industries where products must...
Read More -
Ten Tips for A Successful Use of Total Product Life Cycle (TPLC) Reports
on 2 June 2014 | By Ryan Kasun
The FDA, as part of the transparency initiative, has developed a Total Product Life Cycle (TPLC) report capability on their website (www.fda.gov). The intention behind this new report is to provide existing data to medical device manufacturers that will enable them to...
Read More -
Ten Tips for Successful Integration of Risk Management & Usability Engineering
on 19 May 2014 | By Ryan Kasun
Even though Risk Management and Usability Engineering are clearly two separate processes with unique requirements, they are interdependent and must communicate with each other during the entire product lifecycle. Risk Management identifies the risk of a medical device’s...
Read More -

The Basics of Polymer Analysis: Techniques & Solutions
on 12 May 2014 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
Given the level to which plastics, rubbers, and other materials have impacted modern life, it is easy to see the importance of in-depth polymer analysis. By exploring critical parameters like molecular weight (MW), structure, morphology, and thermal characteristics, chemists can...
Read More -
510(k) Submissions-Ten Tips for a Successful Submission
on 5 May 2014 | By Ryan Kasun
The process of submitting a 510(k), demonstrating that your device is substantially equivalent to a device that is already cleared, can be daunting. Typically designed to be a 90-day review by the FDA, the entire process could take several months if adequate information is not...
Read More -
Successful Regulatory Strategy-Ten Tips
on 21 April 2014 | By Ryan Kasun
Having a documented regulatory strategy developed early during a new product’s development life cycle and updated periodically is critical for a new product’s market success. An inaccurate or lack of regulatory strategy can lead to the product not being cleared for market in the...
Read More -
Implementing a Successful Recall Process - Ten Tips
on 24 March 2014 | By Ryan Kasun
In the medical device arena, recalls are a fact of life and competitors love to find out that you’ve had one. Regardless of recall classification (Class I, II, or III), a recall procedure is instrumental in working in concert with a company’s post-market surveillance and...
Read More -
Warning Letters - Ten Tips
on 10 March 2014 | By Ryan Kasun
Each year, the FDA issues hundreds of warning letters to medical device companies for reasons ranging from manufacturing practice violations to breaches in labeling and misbranding. Companies responding well to the FDA’s demand for prompt compliance will ultimately return to the...
Read More -
Pre-Submission Program – Final Guidance
on 21 February 2014 | By Ryan Kasun
On February 18, the FDA issued the final guidance document for the Pre-Submission Program and Meetings with FDA Staff. The Pre-Submission process is a way for industry to get feedback from the FDA prior to the submission of a premarket application (PMA, 510(k), HDE, IDE, de...
Read More -
Affordable Breakthrough Innovations
on 13 February 2014 | By Ryan Kasun
Innovative ideas for medical devices are often costly for manufacturers to make and for patients to own. However, not all innovative medical devices will cost you an arm and a leg.
Read More -
Guidance Documents for 2014
on 30 January 2014 | By Ryan Kasun
In an effort to help get safe and effective medical devices to market more quickly, the FDA publishes a list of proposed guidance documents each year. Here is FDA’s prioritized list of things you can expect to see in 2014:
Read More -
Pediatric Subpopulations – Final Rule
on 22 January 2014 | By Ryan Kasun
On Friday, January 10, 2014 the FDA issued a final rule, which amends the premarket approval (PMA) regulations. The final rule requires “submission of information on pediatric subpopulations that suffer from the disease or condition that a device is intended to treat”. 1 The...
Read More -
510(k) Pop Quiz!
on 10 January 2014 | By Ryan Kasun
How well do you know the logistics of the 510(k) process? Today’s blog is interactive, as well as educational! Take this quiz (no cheating!). Comment on the post to tell me how many answers you got right!
Read More -
3D Printing in Medicine
on 9 January 2014 | By Ryan Kasun
As if 3D printers for rapid prototyping weren’t cool enough, researchers at the University of Wollongong and St. Vincent’s Hospital Melbourne, part of Australian Research Council Centre of Excellence for Electromaterials Science (ACES), have been using 3D printing to make some...
Read More -
Regulatory News - Report on FDA Device Review Process
on 9 January 2014 | By Ryan Kasun
As part of FDA’s Performance Goals and Procedures adopted under MDUFA III (The Medical Device User Fee Act of 2012), the FDA agreed to participate with the medical device industry in an independent assessment of the process for the review of medical device submissions. The key...
Read More -
Transhumanism - Science from Fiction: Conclusion
on 2 January 2014 | By Ryan Kasun
Trans∙hu∙man∙ism /tranz’hyoomənizm// Noun The belief or theory that the human race can evolve beyond its current physical and mental limitations, especially by means of science and technology.1
Read More -
Devices for the Military
on 20 December 2013 | By Ryan Kasun
This past weekend I travelled to the Chicago-land area to attend my brother’s graduation from Northern Illinois University and commissioning into the US Army. He will be reporting to Fort Leonard Wood, Missouri for officer training as part of the Army Corps of Engineers. This...
Read More -
Veterinary Medical Devices - FDA Regulations
on 12 December 2013 | By Ryan Kasun
I recently worked on a project that involved writing a regulatory strategy for a device used on animals. This was a new area for me and I got interested in learning even more about the topic afterwards. I was curious as to how big of a market veterinary devices were, as well as...
Read More -

Risk Assessment Strategies in Extractables and Leachables
on 6 December 2013 | By Kevin Rowland, Jordi Labs/RQM+ Director of R&D
Risks assessments are a key element in extractables and leachables testing. There are many factors involved in ascertaining the specific risks and how this ultimately impacts the biocompatibility of a medical device.
Read More -

5 Key Elements of GMP in the Food Industry
on 4 December 2013 | By Kevin Rowland, Jordi Labs/RQM+ Director of R&D
Good manufacturing practice (GMP) is a critical part of the quality assurance and control (QA/QC) pipeline for food contact materials, from primary packaging through to kitchenware. Although GMP principles were established by the US Food and Drug Administration (FDA), they were...
Read More -

Getting to Know You
on 2 December 2013 | By Ryan Kasun
You probably think you know yourself pretty well, right? Of course. You’re a (circle all that apply) super cool/ witty/ charming/ good-looking/ intelligent person, and humble about it too, but what does your DNA say about you? DNA is an incredibly complex storage structure for...
Read More -

Molecular Weight Analysis: How to Measure MW?
on 22 November 2013 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
Analyzing the molecular weight of both natural and synthetic polymers is extremely complex, and almost impossible to carry out with exactness. Unlike elemental analysis – which focuses on the chemical composition of samples under test – molecular weight analysis considers the...
Read More -
Devices remain unclassified
on 14 November 2013 | By Ryan Kasun
I read a really interesting article this week written by Josh Rising called "The Loophole That Keeps Precarious Medical Devices in Use". The articles explains how when the Medical Device Amendments of 1976 were implemented, dozens of marketed medical devices were left...
Read More -
A Culture of Quality
on 7 November 2013 | By Ryan Kasun
As quality professionals, we are always looking for ways to improve the quality of products, and reduce the cost of quality mishaps. But how do you actually climb to the top of that mountain of high quality while still maintaining a marketable product?
Read More -

Not-So-Blind Ambition
on 6 November 2013 | By Ryan Kasun
Can you imagine what a GPS for blind people would be like? We may not be there yet, but a research project at the University of Arkansas could be the next best thing. Dr. Cang Ye and his team are making over the standard white cane often used by the visually impaired into a...
Read More -
RAPS 2013 Series: MDSAP
on 31 October 2013 | By Ryan Kasun
Of all of the seminars I attended at the RAPS 2013 Conference in Boston, the Medical Devices Single Audit Program ("MDSAP") was arguably the best. The overview of MDSAP - done well by Kim Trautman of the FDA and Mike Ward from Health Canada - was thorough and well done; however,...
Read More -

Quality Control for Blood Transfusions
on 24 October 2013 | By Ryan Kasun
Over the years, major advances in blood transfusion safety measures have drastically reduced the risk of viral transmission via allogeneic blood. Although correlation doesn’t necessarily imply causation, it’s tempting to believe that this may have something to do with the...
Read More -
First Artificial Pancreas receives FDA Approval
on 24 October 2013 | By Ryan Kasun
Upon my nightly adventures of surfing the World Wide Web I came across this article on the Popular Science website announcing the FDA’s approval of the Medtronic MiniMed 530G Insulin pump, praisingly dubbed the first artificial pancreas. The Medtronic device was officially...
Read More -
Medical Device Manufacturers | Device Manufacturers | Affordable Care Act | Medical Device News | Medical Device Tax
On the Edge of Our Seats
on 18 October 2013 | By Ryan Kasun
As the government shutdown dragged onward, nobody was watching the outcomes more than medical device manufacturers. For device manufacturers – there was and still is a lot at stake. And make no mistake – the medical device tax is part of the hot debate in Washington right now....
Read More -
Deep Breaths
on 18 October 2013 | By Ryan Kasun
The standard treatment for patients with cancer of the larynx is to have a tracheostomy tube installed. Tracheostomy tubes are Class II devices, under 21 CFR 868.5800. According to ProTip CEO Maurice Beranger, this technology has not changed in 140 years. Tracheostomy tubes...
Read More -
Government Shutdown - Impact on Medical Devices
on 15 October 2013 | By Ryan Kasun
On October 1st, 2013, the Government of the United States began shutting down operations due to an impasse in congress regarding government funding appropriations. Practically speaking for the medical device industry, the shutdown puts businesses, medical professionals, and,...
Read More -
Transhumanism - Science from Fiction: Part I - Bionic Body Parts
on 11 October 2013 | By Ryan Kasun
Advancements in biotechnology have made things of science fiction into reality. Back in the 70’s, bionic body parts were thought to be donned only by the Six Million Dollar Man1 and The Bionic Woman2. Fast forward forty years and bionic body parts are more science than fiction....
Read More -
Telehealth
on 11 October 2013 | By Ryan Kasun
In the world in we live, where communication is becoming further removed from face to face interaction, medicine is utilizing telecommunications technologies to advance treatment and care for patients.
Read More -
FDA gives Voice to the People
on 11 October 2013 | By Ryan Kasun
The Center for Devices and Radiological Health (CDRH) at the Food and Drug Administration (FDA) has launched a new initiative targeted at giving the patients a voice in the agency’s decision making process on medical devices. I became aware of this effort by the FDA when...
Read More -

-
Canada | Change In Contract Manufacturer | Health Canada | Medical Device | License Amendment Fax Back Form | Registration Triggers | Registration | Change In Country Of Origin
License Amendment Triggers in Canada
on 2 October 2013 | By Ryan Kasun
Sometimes changes in process can have a dramatic effect on product registration. There are several events that can cause a re-registration of a registered medical device. A new product name, part number, material, manufacturing site, country of origin, business name or...
Read More -
Pounding the Pavement
on 1 October 2013 | By Ryan Kasun
Earlier this year I joined a running club, Steel City Road Runners (come run with us!). I’m still a newbie, but I really enjoy the challenge that running provides. This past week, the Boston Marathon announced to applicants who would be awarded the last 5,000 spots in the 2014...
Read More -
-
FDA Releases Mobile Medical Applications Guidance Document
on 27 September 2013 | By Ryan Kasun
This week must be the week of the FDA publications. In the same week the UDI final rule and guidance were published, the FDA has also released the guidance document on the FDA's plan for enforcement regarding Mobile Medical Applications. The FDA's final thoughts on how they'll...
Read More -
UDI Documents Released by FDA
on 25 September 2013 | By Ryan Kasun
The rule and guidance document for Unique Device Identifiers ("UDI") on medical devices and some combination product that contain devices has been released.
Read More -
EU marketing approval system
on 19 September 2013 | By Ryan Kasun
The European marketing approval system for medical devices is less stringent than other regions of the world and because of that, at times it is viewed as a more attractive market for device manufacturers. However over the past few months, a new proposal has come up in Europe...
Read More -
Same Road (Regulatory Pathway), Different Car (Medical Device)..Similar Success (Clearance,Approval)
on 18 September 2013 | By Ryan Kasun
As you might expect, here at RQS the responsibilities and duties of providing our clients with the support they need often involves traveling for on-site visits. For the time being, most of the travel takes place within the greater metropolitan area of the cities in which we...
Read More -
Transhumanism - Science from Fiction: Intro
on 17 September 2013 | By Ryan Kasun
What do Jean Grey and Professor Xavier of X-Men, Vulcan “mind-melds” from Star Trek, the Six-Million Dollar Man, Darth Vader and Jedi mind tricks from George Lucas’ Star Wars, The Bionic Woman adapted from Martin Caidin’s Cyborg, Inspector Gadget, Replicants from the 1980’s...
Read More -

A Systematic Study of Response Factor Variation for Extractables and Leachables
on 11 September 2013 | By Kevin Rowland, Jordi Labs/RQM+ Director of R&D
Chemical analysis for leachables is one of the the primary methods used to assess the risk to patients and consumers from substances which leach from pharmaceutical packaging, medical devices and food packaging. A number of high-profile incidents have demonstrated that...
Read More -
International Labeling Requirements
on 11 September 2013 | By Ryan Kasun
It used to be much easier to have an international device on the market that complies with the regulations of the multi-country international market. With a properly localized multi-language Instructions for Use, the regulations of most international countries were met with one...
Read More -
Brain Disease | Iso 14971 | Risk Management | Harm | Sequence Of Events | Hazardous Situation | Sterilization | Hazard
Foreseeing the Risks
on 5 September 2013 | By Ryan Kasun
On the news this morning, I heard a story that got me thinking about risk management. The headline was “Eight N.H. patients possibly exposed to fatal brain disease”. Further research about the situation reveals just how complex this case is.
Read More -
510(k)-Exempt Devices
on 5 September 2013 | By Stephen Biernacki
In the FDA-regulated medical device world, there are 3 classifications for medical devices: Class I, Class II and Class III. The FDA (http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/Overview/ClassifyYourDevice/default.htm) provides a rather in-depth overview of how...
Read More -

-
FDA guidance document on RF Wireless Technology in Medical Devices
on 30 August 2013 | By Ryan Kasun
With the growing use of incorporating wireless technology into medical devices, the FDA has issued a final guidance Radio Frequency Wireless Technology in Medical Devices. This guidance document is meant to cover a wide range of medical devices that use wireless technology from...
Read More -
Improved machines based on human biology
on 30 August 2013 | By Ryan Kasun
Today, when I was thinking of a blog topic, I had a moment of clarity. I realized that I regularly peruse the news and media for evidence of how machines and medical devices help humans, improve human life, and how humans utilize machines and technology to help solve problems. I...
Read More -
Recognition for Contributions in Medical Advancement 60 years in the making
on 30 August 2013 | By Ryan Kasun
In my Bioethics course during my undergraduate years, we had discussed the curious case of Henrietta Lacks: a 31 year old African American mother who contributed to numerous modern day medical advancements upon her death without her or her family’s knowledge. I noticed that...
Read More -
Reliability – From Bike Racks to Medical Devices
on 22 August 2013 | By Ryan Kasun
Every once in a while, when I’m not running, I like to take my bike down to the river trails of downtown Pittsburgh. I throw my bike rack on my car, strap it in, and I’m ready to go. Being as I only used my bike rack about ten times since I got it, I was surprised when I went to...
Read More -

Vacation Revelation
on 22 August 2013 | By Ryan Kasun
I am in TopSail North Carolina this week enjoying the ocean, sun and family! (Yes: in that order)
Read More -
Class Iia | Eu | Disease Management | Mobile Platform | Mobile Medical Applications | Health Management | Fda | Substantial Equivalence | Verizon Wireless | Software Platform | Medical Applications | Software Technologies | Medical Application
Mobile Medical Applications - Most Reliable Apps, Internationally?
on 22 August 2013 | By Ryan Kasun
A few notable posts pertaining to mobile medical applications came through my Twitter feed / Inbox recently, so I wanted to share!
Read More -

Fear Factor: Audits
on 22 August 2013 | By Ryan Kasun
Why is it that when you drop the A-bomb (audit) people just seem to instinctively cringe. Is this a learned behavior or one that has been passed down since men began assessing each other for acceptance?
Read More -
RQS Ohio Social Event - August 14, 2013
on 9 August 2013 | By Ryan Kasun
Please join RQS for a happy hour networking event! Drinks and appetizers will be provided and prizes will be given away to winning attendees during the event.
Read More -
Guidance document clean-up! - CDER
on 9 August 2013 | By Ryan Kasun
I read an interesting article on the RAPS website this morning about a new initiative set by the FDA to clean-up certain guidance documents. This means finalizing draft guidance documents, withdrawing or updating/revising outdated guidance documents. This initiative is taking...
Read More -
Draft Guidance on Medical Device Reporting for Manufacturers - Issued 07/09/13
on 5 August 2013 | By Ryan Kasun
Earlier last month, FDA released a new draft guidance that addresses many questions and answers pertaining to the sometimes tricky world of medical device reporting for medical device manufacturers. (1-4) The draft guidance, “Draft Guidance on Medical Device Reporting for...
Read More -
Super Supplements or Scary Supplements: Consumers are the Judge
on 1 August 2013 | By Ryan Kasun
While doing my usual read-through of the headlines on USA Today, I recently came across an article about a USA Today investigation examining the works of Matt Cahill, a supplement designer with a dark history in developing risky products. As an individual who tries to maintain a...
Read More -
“It’s like getting your high school diploma…”
on 26 July 2013 | By Ryan Kasun
Recently, I had the opportunity to attend a Risk Management Conference hosted by the FDAnews. At the beginning of the conference, we all went around the room and introduced ourselves, and indicated why we were there. Not surprisingly, a majority of people were there to have a...
Read More -
Just Say No
on 24 July 2013 | By Ryan Kasun
So you think you’re a bargain shopper, huh? Did you secretly get a rush when you had a coupon for that high-end toothpaste AND found it on sale? Most bargain shopping comes with few, if any, downsides. (Ok, so maybe you had to evict a family of angry possums from that couch you...
Read More -
FDA Medical Device Reporting Update
on 24 July 2013 | By Ryan Kasun
One of the ways that I try to stay in touch with the world of medical device regulations is by subscribing to the CDRH (Center for Devices and Radiological Health) mailing lists (http://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CDRH/ucm135906.htm)....
Read More -
How the FDA handles complaints
on 19 July 2013 | By Ryan Kasun
Complaints from end users to medical device manufacturers are unfortunately a common occurrence. Medical device manufacturers are expected to handle these complaints efficiently and effectively by keeping the end user or patient’s safety in mind. However not a lot of people...
Read More -
Biosense | 21 Cfr | Fda Guidance | Mobile Medical Applications | Fda | Medical Device | Submission Type | Mobile Application | Medical Applications | Regulatory Strategy | Medical Application
Mobile Medical Applications: Make Sure uChek with FDA
on 19 July 2013 | By Ryan Kasun
The latest mobile medical application news on the industry's collective mind is the issuance of an "It has come to our attention..." letter sent to Biosense Technologies in India, essentially questioning why Biosense has not obtained 510(k) clearance for their uChek mobile...
Read More -
Documentation 101: Life as an Engineer
on 11 July 2013 | By Ryan Kasun
As a recent college graduate working in the medical device industry, I quickly learned that life as an engineer is different than what is envisioned and different than what is taught in the hundreds of undergraduate programs across the nation. Today, students graduate with...
Read More -
Croatia Joins the EU
on 10 July 2013 | By Ryan Kasun
As of June 30th, Croatia has now been formally added to the list of EU governed / regulated countries making for a total of 27 European Union countries.
Read More -
A Vaccine for Colon Cancer
on 10 July 2013 | By Ryan Kasun
As a University of Pittsburgh graduate, I was excited to read in the spring issue of Pitt Magazine about a promising advance in cancer prevention research at my alma mater. Immunologist Olivera Finn and her research team at Pitt have been studying potential cancer prevention...
Read More -
The Importance of Production Equivalence
on 10 July 2013 | By Ryan Kasun
In today's world of always looking for ways to get something done faster there appears to be one area where speed has begun to do some harm. This area happens to be related to production equivalence. Quite simply, production equivalence is the evidence that a manufacturer...
Read More -

What are Extractables and Leachables?
on 3 July 2013 | By Mark Jordi, Ph.D., RQM+ Principal Advisor
Container systems for drug products should not release any chemicals which can build up in quantities that could create a risk of toxicity or impact the stability or efficacy of the drug. Extractables and leachables refer to compounds which can be either extracted or leached...
Read More -
Cybersecurity | Premarket Submissions | Risk Analysis Approach | Patient Data | Security Breach | Mobile Apps | Medical Device Design
Cybersecurity
on 28 June 2013 | By Ryan Kasun
My recent work has involved providing regulatory assistance to software medical devices. One thing I have been learning about is how security and privacy is handled with these types of products. Often times I have experienced clients grouping regulatory affairs with privacy and...
Read More -

My Day at FDA
on 28 June 2013 | By Ryan Kasun
For today's show-and-tell, I'm bringing a picture of me at FDA:
Read More -
FDA Inspections Database
on 27 June 2013 | By Ryan Kasun
The Food and Drug Administration (FDA) recently released its new data set for the Inspections Database. 1-2 The Inspections Database makes available the most recent inspection (up to two years of inspections) of a company. The final inspection classification for clinical trial...
Read More -
Start Ups | Vcs | Due Diligence | Investments | Angel Investors | Exit Strategy | Venture Capitalists | Venture Capitalist | Start-up Device | Medical Device Start-up | Angel Group
Funding Your Start-Up Medical Device Company
on 27 June 2013 | By Ryan Kasun
Over the last few months, several of the potential clients I’ve met with have been start-up device companies. Typically, I find that start-ups will have a fantastic product idea that can positively impact countless lives – but, often, they do not have a clear plan for how they...
Read More -
Congenital Heart Defects | Rqs | Heart To Heart | Medical Technology | Quality Solutions | Birth Defect | North Park
Congenital Heart Walk
on 19 June 2013 | By Ryan Kasun
This weekend my family and I are going to go to the Congenital Heart Walk at 9:45 a.m. at the boathouse in North Park. This walk is to raise awareness for Congenital Heart Defects, America’s #1 birth defect. Regulatory and Quality Solutions is a sponsor of this event; it is very...
Read More -
Software System | Regulatory Focus | Www.fda.gov | Fda | Fda Website | Maude Database | Software | Regulatory | Graematter | Regulatory Intelligence
Regulatory Intelligence
on 14 June 2013 | By Ryan Kasun
How much time to you spend searching FDA’s website? Looking for predicates, collecting adverse event reports from the MAUDE database, searching for guidance documents, etc. I’m sure I don’t need to dig into the details, you have felt this pain too if you have spent any...
Read More -
Eye Spy
on 14 June 2013 | By Ryan Kasun
Every time I visit my optometrist I see advertisements for colored contact lenses. I’m pretty happy with the green I’ve got so I’ve never tried another color. Did you know that contact lenses, even the purely decorative ones that don’t correct your vision, are considered medical...
Read More -
Consumer Medical Device Adverse Event Reporting
on 14 June 2013 | By Ryan Kasun
Recently I went trolling through the FDA website and stumbled upon an article entitled "Wanted: Consumers to Report Problems (http://www.fda.gov/ForConsumers/ConsumerUpdates/ucm354560.htm#report). It's a great article that at its core, is a plea to the consumers of medical...
Read More -
Integration Phase - AGILE vs. Regulatory
on 14 June 2013 | By Ryan Kasun
An AGILE software development process allows for adaptive development through incremental changes in the software. This means that there are more releases and upgrades of the software in the field. Within the AGILE process there is time allotted for the integration phase. This...
Read More -

Race for the Place and RQS Cares
on 6 June 2013 | By Ryan Kasun
After reading Steve’s blog, "Why RQS Cares- The Race for the Place" and learning more about Race for the Place/The Gathering Place, I knew that I wanted to participate in this event. Soon thereafter, I was registered and ready to participate in RQS Cares’ first Ohio-based...
Read More -
Submissions | Fda | Medical Device | Team Members | Regulatory | 510(K) | Approval Time | Medicaldevice | Customer Success | Collective Experience | Regulatory Professionals | Fda Clearance
510(K) Success!
on 3 June 2013 | By Ryan Kasun
RQS team members pride ourselves on focusing on customer success, and it is with that in mind that I am honored to announce our most recent 510(K) submission was approved this week! We are proud to obtain clearance, but swell with pride because of how we obtained it.
Read More -
Mobile Medical Applications - More Apples, More Hassles
on 29 May 2013 | By Ryan Kasun
Could it get much worse for mobile medical applications and their developers trying to utilize phones and tablets as point-of-care medical devices? OK, surely it could, but this story can't make it any easier.
Read More -
FDA 510(k) Memorandum #K97-1 “Deciding When to Submit a 510(k) for a Change to an Existing Device
on 29 May 2013 | By Ryan Kasun
The FDA is looking for industry input for the revision of FDA 510(k) Memorandum #K97-1 “Deciding When to Submit a 510(k) for a Change to an Existing Device,” January 10, 1997. This is a critical go-to document for the average Regulatory Engineer, so I have been thinking about...
Read More -
Medical Device Report | Complaint Handling | Medwatch | Complaints | Fda | Medical Device | Mdr | Adverse Event | Post Market Vigilance
Complaint Handling Solutions
on 23 May 2013 | By Ryan Kasun
In my last post “Who’s watching your MedWatching”, I commented on some of the common issues manufacturers have with their MDR and complaint handling systems. It’s useful to know what people are doing wrong, but I think it is equally important to know how to solve those issues....
Read More -
Medical Cybercrime
on 23 May 2013 | By Ryan Kasun
I saw an episode of Elementary a couple weeks ago in which a hired assassin killed someone by hacking into his pacemaker and cranking up the voltage to cause a heart attack. Initially I thought maybe crime shows were just stepping up their game, trying to find more creative ways...
Read More -
Internal Audits, the After-Thought of a Quality System
on 23 May 2013 | By Ryan Kasun
Why is it that Internal Audits are forgotten, or even worse feared? Why is it so many organizations of today place so much focus on the creation of Quality Procedures, Work Instructions, Forms, etc. but do not place the same focus on conducting internal audits of the quality...
Read More -
Using AGILE in Medical Device Software Development
on 17 May 2013 | By Ryan Kasun
In school I was always taught that the medical device development process followed the waterfall model. However since entering the work force I have started to see other methods of development for different types of devices. I have recently been working with software medical...
Read More -
MedCon 2013
on 16 May 2013 | By Ryan Kasun
At the beginning of this month, MedCon 2013 was hosted in Cincinnati, OH by Xavier University. The three day conference was held Wednesday May 1st, 2013 through Friday May 3rd, 2013.
Read More -
 Low Heart Rate | Congenital Heart Defects | Congenital Heart Defect | Heart Block | Pacemaker | Adult Congenital Heart Association | Rqs Cares | Heart Foundation
Low Heart Rate | Congenital Heart Defects | Congenital Heart Defect | Heart Block | Pacemaker | Adult Congenital Heart Association | Rqs Cares | Heart FoundationWhy RQS Cares - Congenital Heart Walk
on 13 May 2013 | By Ryan Kasun
Today we feature a post from Deb Gimbel on why RQS Cares has chosen to support the Congenital Heart Walk. Seven years ago, my niece Riley was born with a congenital heart defect known as complete heart block, which is an interference in the signals between the heart chambers,...
Read More -
Localization Issues
on 9 May 2013 | By Ryan Kasun
Last month Serbia changed its localization so that The Instructions for Use and labeling of medical devices must be written in the Serbian language. There was no grace period for this change, it was effective immediately. Conversely, Korea has also indicated that their...
Read More -
Training, a Test of Endurance
on 8 May 2013 | By Ryan Kasun
I was blessed enough this weekend to run the Pittsburgh Half-Marathon with my wife, her dad, and her cousin. I had such a blast running in my favorite city in gorgeous weather with 30,000 other crazy people!
Read More -
 Cancer | Gathering Place | Diagnosis Treatment | Race For The Place | Cancer Survivor | Brain Cancers
Cancer | Gathering Place | Diagnosis Treatment | Race For The Place | Cancer Survivor | Brain CancersWhy RQS Cares - The Race for the Place
on 6 May 2013 | By Ryan Kasun
Today we feature a post from Steve Keverline on why RQS Cares has chosen to support The Race for The Place. My father, sister and I stood in the hospital room by my mother’s side. We were anxious to learn the cause of her recent dizzy spells and loss of appetite. She had a CT...
Read More -
When Will Academia Catch-up to the Needs of the Medical Device Industry?
on 3 May 2013 | By Ryan Kasun
This past weekend I had a really great discussion with some neighborhood friends that really didn't know much of my background (where had I grown up, gone to school, etc.) and the question came up "what did you study in college?" This is a question that I'm sure everyone has...
Read More -
Medical Device Report | Complaint Handling | Warning Letter | Medwatch | Complaints | Fda | Medical Device | Mdr | Adverse Event | Post Market Vigilance
Who’s watching your MedWatching?
on 3 May 2013 | By Ryan Kasun
Last year, FDA received almost one million adverse event reports from manufacturers1. Needless to say, the Office of Surveillance and Biometrics has been very busy, and so have manufacturers.
Read More -
Everyday Usability – Alarm Clock Edition
on 1 May 2013 | By Ryan Kasun
I’ve always had a rocky relationship with my alarm clock, specifically the snooze button. In my current clock it’s built into the base (not the display, where all the cool buttons hang out), and it’s angled, narrow and difficult to locate in a semi-conscious state. I pretty much...
Read More -
Biocompatibility | Medical Devices | Premarket Submissions | G95-1 | Standard Iso | Iso 10993-1 | Fda | Draft Guidance Document | Guidance Document | Biological Evaluation | Iso 10993 | Fda Clearance
New Biocompatibility Draft Guidance
on 25 April 2013 | By Ryan Kasun
FDA issued a new draft guidance document on biocompatibility on Tuesday April 23, entitled Use of International Standard ISO-10993, Biological Evaluation of Medical Devices Part 1: Evaluation and Testing. It is intended to replace the ODE General Program Memorandum #G95-1. G95-1...
Read More -
Harmonized Symbols | Medical Devices | Medical Device Labels | Standards Development Organization | Labeling | Regulators | Labeling Products | Fda | Medical Device | Labels | Regulatory Structure | Biological Product | Sdo
Crash Those Symbols - FDA Proposed Rule
on 24 April 2013 | By Ryan Kasun
Make some noise people - the FDA may be making labeling easier on the medical device and biological product industries by way of harmonized symbols!
Read More -
How We Conduct ISO 10993 Medical Devices Evaluations
on 23 April 2013 | By Kevin Rowland, RQM+ Director of R&D
The goal of ISO 10993 is to protect patients from biological risks arising from the use of medical devices. The standard covers testing for genotoxicity, carcinogenicity, cytotoxicity and a variety of other areas of concern. While the standard covers specific areas of testing to...
Read More -
Shock | Fda | Medical Device | Tachycardia | Fibrillation | Electrodes | Sudden Cardiac Arrest | Cardiac Emergency | Electrode Pads | Heart Rhythms | American Red Cross | Aeds
Proposed Order on AEDs
on 23 April 2013 | By Ryan Kasun
Automated external defibrillators (AEDs) can save the lives of individuals suffering from a cardiac arrest event. Sudden cardiac arrest can happen to anyone and at any time. According to the American Red Cross, over 350,000 people will suffer from sudden cardiac arrest this...
Read More -
Congenital Heart Walk | Children's Heart Foundation | Fundraising Event | Rqs | Congenital Heart Disease | Chd | Acha | Promising Research | Heart Foundation | Heart Disease
RQS Cares - Congenital Heart Walk
on 22 April 2013 | By Ryan Kasun
RQS is happy to announce that RQS Cares is organizing a group for the Congenital Heart Walk event occurring on June 22nd, 2013 in Allison Park, PA. The race serves as a community gathering and fundraising event for ACHA, an organization to improve the quality of life and extend...
Read More -
RQS Cares - Race for the Place
on 22 April 2013 | By Ryan Kasun
RQS is happy to announce that RQS Cares is organizing a group for the Race for the Place 5K run / 1 mile walk event occurring on June 2nd, 2013 in Beachwood, OH. The race serves as a fundraising event for The Gathering Place - an organization focused on supporting, educating,...
Read More -
Quality Management | Medical Device Companies | Corporate Quality | Management Reviews | Quality System | Quality Manager | Corporate Policy | Manufacturing Process | Executive Management Team | Quality Policy
Quality Systems Becoming the Culture of a Company
on 18 April 2013 | By Ryan Kasun
The culture of a company is what drives the organization – it can change and adapt, but ultimately, if a strong management team doesn’t drive a company’s culture – the company risks losing its identity. For medical device companies, establishing a culture around the Quality...
Read More -
Hpfb | Medical Device Companies | Wikipedia | Money | Tga | Regulatory Risk | Medical Device Industry | Investors | Fda | Medical Device | Anvisa | Engineering Projects | Open Source Projects | Crowd Source
Crowd Source in Medical Devices?
on 17 April 2013 | By Ryan Kasun
Collaborative development, or for the purposes of this post we'll call it "crowd sourcing", seems to be discussed and used more frequently in the current business environment. Obviously, crowd sourced projects have been around for a long while (Wikipedia is just one well-known...
Read More -
Mammogram | Medical Devices | Mammograms | Usability | Medical Device | Breast Cancer Screening | Human Factors | Regulatory | Women In Bio | Quality | Breast Cancer
Human Factors
on 16 April 2013 | By Ryan Kasun
Recently I had to get my annual mammogram. They were running behind like doctor’s offices sometimes do - there were seven women in dressing gowns in the waiting room with me. After a few awkward moments, we all started to talk to each other. Every single one of the ladies talked...
Read More -
Investors | Patent | Patent Application | First To File | Medical Device | Patent Office | Law | Innovation | First To Invent | Us Patent Law | Patent System
Patent Law Changes
on 11 April 2013 | By Ryan Kasun
For over 200 years, the US has operated under a “first to invent” rule. This means that if two innovators independently submit a patent for the same exact invention, the person who can prove that they invented it first is the one who is awarded the patent.
Read More -
Ergonomics Society | Alarms | Usability | False Positives | Alarm Systems | Medical Device | Iec 60601 | Human Factors
An Alarming Trend
on 10 April 2013 | By Ryan Kasun
If you’ve ever spent any appreciable amount of time in a hospital or other medical care facility, you may have been driven temporarily insane by the never-ending chorus of medical device alarms. I was recently reminded of a few nights I spent in the hospital with my dad a year...
Read More -
FDA's Guidance of User Fees and Refunds for Premarket Notification Submissions 510(k)s
on 10 April 2013 | By Ryan Kasun
This morning I was welcomed by receiving an email from the FDA CDRH News stating that new guidance was released regarding user fees and refunds for pre-market notification submissions 510(k)s. Some of the takeaways I had were:
Read More -
FDA’s Guidance Document on Voluntary Submission of ISO 13485 3rd Party Audit Reports
on 5 April 2013 | By Ryan Kasun
I came across an FDA guidance document – not entirely recent; however, I found it very interesting and worth sharing. It is on voluntary submissions of audit reports done by auditing bodies outside of the FDA.
Read More -
Audit Training
on 5 April 2013 | By Ryan Kasun
I recently had the opportunity to receive some training in auditing of medical device companies. In this training I was able to gain experience from an auditor's perspective as well as an auditee's perspective on how an audit is performed. This training was important to me...
Read More -
Trade | Mdd | Medical Devices | Trade Deadline | Trade-off | Nhl | Iso 14971 | Risk Management | 14971 | Fda | Business Risk | Cmdr
Medical Device Development: Trades
on 3 April 2013 | By Ryan Kasun
As a somewhat rabid hockey fan there are a few days during the year that are considered sacred holidays: every playoff game, first day of free agency, and the trade deadline. My mind is on hockey because the trade deadline happens to be today. Outside of my insatiable desire to...
Read More -
Excise Tax | Budget Resolution | Bipartisan Vote | Medical Device Manufacturers | Us Senate Committee | Senate Committee On Finance | U S Senate | Fda | Medical Device | Bipartisan Support | Government | Affordable Care | Medical Device Tax | Bipartisan
Bipartisan Repeal of Medical Device Excise Tax
on 2 April 2013 | By Ryan Kasun
Bipartisan support, in today’s world of politics, might seem a little hard to believe. However, the U.S. Senate passed an amendment that repeals the medical device tax last month (Thursday, March 21, 2013). The bill was passed by a bipartisan vote of 79 to 20. 1 This impressive...
Read More -
Medical “Wow”: 3D Printed Skull
on 28 March 2013 | By Ryan Kasun
Last week I saw several articles published about a new device that is going to shake up the Craniomaxillofacial device market, and the Orthopedic market is next on the list. 3D Printing – chances are you’ve just started hearing about it, mostly concerning gun controls laws for...
Read More -
March Madness
on 26 March 2013 | By Ryan Kasun
The season is upon us. Unfortunately, the season is not spring…winter still seems to be lingering. However, the season of March Madness is in full swing! Everyone’s bracket selections are in, and we wait to find out who is the National Champion.
Read More -
Correction | Medical Devices | Warning Letter | Fda Inspection | Immediate Action | Fda | 483 | Medical Device | Corrective Action
RQS Feature Article - Successfully Addressing Warning Letters
on 22 March 2013 | By Ryan Kasun
Regulatory and Quality Solutions is featured in another publication this week! Our President, Maria Fagan, has written an article focused on the top five tips on how to successfully address FDA warning letters. The article is featured in MedCity News and can be found here. FDA...
Read More -
Medical Devices | Ergonomics Society | Medical Device Industry | Human Factors Standards | Human Error | Fda Perspectives | Industry Guidance | Regulatory Submissions | Usability Engineering | Cdrh | Human Factors Group
HFES Health Care Symposium – What’s New in Human Factors and Usability?
on 20 March 2013 | By Ryan Kasun
Last week I had the opportunity to attend the Human Factors & Ergonomics Society Health Care Symposium. Since I work on medical devices, I mainly went to the presentations in the medical devices track and found them to be very interesting and educational. The opening keynote...
Read More -
Quality Agreement | 21 Cfr | Supplier Selection | Medical Device Industry | Fda | Supplier Management | Supplier Quality
Getting Control of Supplier Controls
on 20 March 2013 | By Ryan Kasun
It seems that for one reason or another supplier controls are a requirement that slips through the cracks of even some of the largest medical device organizations. Those of us in the medical device field should know better since 21 CFR Part 820 clearly includes requirements that...
Read More -
RQS appears in TEQ Magazine
on 15 March 2013 | By Ryan Kasun
In the March 2013 edition of the Pittsburgh Tech Council's TEQ Magazine, Jonathan Kersting highlights Regulatory and Quality Solutions' growth over the past 5 years. See here for the blog post that recounts the article. The "e-zine" can be found here.
Read More -
Alternative Materials | 21 Cfr | Leachables | Human Exposure | Dehp | Natural Rubber Latex | Latex | Medical Device Industry | Exposure Levels | Allergy | Materials | Bpa | Phthalate | Polyvinyl Chloride Pvc | Ethylene Vinyl Acetate | Phthalates
Latex and DEHP and BPA, Oh my!
on 15 March 2013 | By Ryan Kasun
They’re just as frightening as Lions and Tigers and Bears, but how much do you know about them? Are they in your medical devices?
Read More -
Language Development | Medical Devices | Cochlear Implant | Medical Technology | Cochlear Implants | Fda | Bionic Eye | Implant Device
Make Sense
on 14 March 2013 | By Ryan Kasun
My son has a friend that was born deaf. When he was a small child, he had surgery to get cochlear implants. With the implants, he is able to hear. When his younger brother was also born deaf, his parents found that they carried a recessive gene that causes deafness. Both boys...
Read More -
Synthetic Rubbers | Latex Allergy | Health Care Industry | Natural Rubber Latex | Food And Drug Administration | Draft Guidance Document | Food And Drug Administration Fda
FDA issues draft guidance for accurate labeling on medical products that are not made with natural rubber latex
on 11 March 2013 | By Ryan Kasun
What do adhesive bandages, blood-pressure monitoring cuffs, sanitary napkins, ventilator bellows, wheel chair cushions, and dental dams all have in common?
Read More -
Standards | European Commission | Ieee | Mdd | Medical Devices | Bsi | Medical Device Field | Medical Device Manufacturer | Country Code | Fda | Astm | Rtca
Keeping Up With Regulations
on 5 March 2013 | By Ryan Kasun
In the medical device field the name of the game is Compliance. The reason for V&V testing is compliance. Prior to distribution you need to provide evidence of compliance. Everywhere you turn there are rules where the medical device manufacturer or distributor must provide...
Read More -
Best Practices | Quality System | Autonomy | Guidance Document | Innovation | Regulatory | Improvements | Quality
Autonomy with Boundaries
on 4 March 2013 | By Ryan Kasun
I have a teenager that just passed his Driver’s Test. As he pulls out of our driveway, I see the freedom, the possibilities, the trouble that he could get into. Although a bit poetic and a bit melancholy, it’s also a bit exciting. His father and I cautiously trust him – he’s a...
Read More -
Tax Payer Dollars | Medical Device Manufacturers | Fda Budget | New Medical Devices | Fda | Advamed | Improved Products
FDA Budget Cuts This Week
on 1 March 2013 | By Ryan Kasun
This week, the FDA faces between 5.1 to 5.3% of their budget being cut this week ($210 million of a $4.1 billion budget). I’ll be interested to see what areas of the FDA are impacted by these budget cuts, and what that means for medical device manufacturers. Proposed to be...
Read More -
Device Submissions | Medical Device | American Medical Association | Pubmed | Guidance Document | Pma Supplement | Medical Product | Draft Guidance | Journal Of The American Medical Association
Open Comments are Not Left Unheard!
on 26 February 2013 | By Ryan Kasun
Some may think the FDA does not take what industry has to say to heart or feel that there is no point in commenting on a proposed rule, because their feedback is left unread.
Read More -
Medical Devices | Collaboration | Medical Device Companies | Partnership | Regulatory Science | Regulatory Pathway | Nih | Fda | Mdic | Medical Device | Innovation | Fda Regulated Products | Medical Device Innovation | Governing Bodies | Innovations
Regulatory Science
on 26 February 2013 | By Ryan Kasun
I heard an interesting new term today (new to me at least!) that I thought greatly impacts the line of work I am in. Regulatory science is "the science of developing new tools, standards and approaches to assess the safety, efficacy, quality and performance of FDA-regulated...
Read More -
Da Vinci | Medical Device Industry | Technology | Medical Device | Technology Advancements | Surgery System | Robot-assisted | Intuitive
Technology Overkill?
on 22 February 2013 | By Ryan Kasun
Over the past few years, there have been some really great technologies that have changed the way we live our lives. Smartphones are the easiest example. Life would be a lot different if I didn’t have instant access to my e-mail, the weather, the spinning schedule at my gym, or...
Read More -
Go Ahead, Take a Chance
on 20 February 2013 | By Ryan Kasun
Throughout our daily lives, we assess risk. At the grocery store, driving to work, giving advice to our kids and buying a car are all examples of internal risk analyses. How can we be sure that we’ve considered everything? Do we know that we’ve given every aspect the proper...
Read More -
Adverse Events | Medical Devices | Quality Engineering | Regulatory Affairs | Design Quality | Mhra | Risk Management | Medical Device | Maude Database | Design Dossier
Regulatory Thoughts
on 19 February 2013 | By Ryan Kasun
Up until the time when I joined the RQS team in November all I had ever truly worked on was Quality. Coming out of college I worked for a plastics bottling manufacturer in process development and quality control. My next job was design V&V closely followed by design quality...
Read More -
New Medical Devices | Medical Device Industry | Medical Device | Regulation | Sensors | Innovations | Medical Breakthroughs
New Medical Devices Coming Soon to a Hospital Near You
on 18 February 2013 | By Ryan Kasun
Recently I came across a FoxNews article (‘Medical breakthroughs on the horizon for 2013’ by Dr. David Samadi) that got me excited. The reason why I pursued the medical device industry was to be in a field that impacted people; a field that matters. I don’t care who you are –...
Read More -
Cleveland Rocks… its way to the top five growing life science hubs
on 12 February 2013 | By Ryan Kasun
After many years of adamantly claiming not to have a “hometown,” hating the 100% humidity-filled summers (that I still despise), and trying to be indifferent about anything “Southern,” I have finally come to peace with my Southern roots and my “Southernisms.” It wasn’t until I...
Read More -
Curling Nationals!
on 12 February 2013 | By Ryan Kasun
Something very exciting is happening this week in Green Bay, Wisconsin. The 2013 Men's and Women's Curling Nationals are taking place all week. What's even more exciting is that this year the semi-final and final games will be on national television. In the past there has only...
Read More -
Qms | Mdd | Eu | Canada | Medical Devices | Fda | Health Canada | 21 Cfr 820 | Preventive Action | Iso 13485
A QMS Outcast: Preventive Action
on 8 February 2013 | By Ryan Kasun
We in the medical device industry throw the term CAPA around like it's a word that is common-place in our society (though, here in Pittsburgh, you'll get a lot of confused looks if you tell someone you've opened a CAPA as it is a local performing arts school. That's a strange...
Read More -
Making Sense of Standards
on 7 February 2013 | By Ryan Kasun
You may have heard that last year there were two new releases of EN standards: EN ISO 14971:2012 and EN ISO 13485:2012. What does this mean to you? What are the changes? How much does this affect the way you do business? Compliance to standards can be somewhat overwhelming....
Read More -
Medical Device Industry | Taxes | Fda | Medical Device | Public Health | Govenment | Regulatory | 510(K) | Cdrh
Headaches
on 7 February 2013 | By Ryan Kasun
The FDA received funding to increase its workforce at the CDRH for 2012 and 2013. They have set aggressive goals to improve the turnaround times for 510K approval. They have instituted programs to retain their employees and to increase the reviewer to manager ratio. My...
Read More -
Auto Compliance
on 5 February 2013 | By Ryan Kasun
Coming to your commercial break at the 2014 Super Bowl at Met-Life Stadium - the automated compliance robot!
Read More -
Internal Auditor | Auditee | External Auditor | Compliance | Observation | Preventive Action | Internal Audits | Corrective Action
Thoughts About Auditing
on 4 February 2013 | By Ryan Kasun
'Tis the season for auditing. The beginning of this year, just like years in the past, seems to have brought the New Year's Resolution that audits will be scheduled and conducted to correct the sins of the previous year and take a fresh look at the upcoming year. To this point...
Read More -
ICH Q1B: Photostability Testing of New Drug Substances and Products
on 31 January 2013 | By Kevin Rowland, RQM+ Director of R&D
The ICH Q1B Guideline was finalized in November 1996. This guidance details the principles required to evaluate the light sensitivity and stability of new drug substances and products. Q1B has been adopted/published/implemented by noteworthy institutions including EC (Europe),...
Read More -
Quality Systems | Submissions | Guidance Documents | Good Manufacturing Practices | Fda | Biological Product | Current Good Manufacturing Practices | Combination Product | Combination Products
Would you like fries with your Combo meal?
on 31 January 2013 | By Ryan Kasun
On January 18, 2013, the FDA announced their final rule on current good manufacturing practices for Combination products. These Guidance Documents will tell companies when submissions would be required for any post-approval changes to a combination product. First off, what are...
Read More -
Final Rule - Combination Products
on 30 January 2013 | By Ryan Kasun
Last week, the FDA issued a final rule(1-2) on combination products(3) and the applicable current good manufacturing practice (CGMP) requirements. The final rule actualizes the proposed rule that was issued many moons ago, in September 2009 without significant changes. Four...
Read More -
Acceptance and Filing Reviews for Premarket Approval Applications (PMAs)
on 28 January 2013 | By Ryan Kasun
In addition to the Refuse to Accept Policy for 510(k)s Guidance document I discussed in my previous post, the FDA also issued a guidance titled Acceptance and Filing Reviews for Premarket Approval Applications (PMAs) at the same time.
Read More -
Iphone | Mhealth | Mobile Medical Applications | Healthspot | Android | Fda | Medical Device | Apps | Health Spot | 510(K) | Retinal Exam
Mobile Medical Applications - Where do we go from here?
on 28 January 2013 | By Ryan Kasun
With the recent announcement of 510(k) clearance of a retinal examining attachment and companion app can we now declare the outcry against the FDA finished?
Read More -
Who is your customer?
on 25 January 2013 | By Ryan Kasun
My mom works as an x-ray technician at a small orthopedic office in Chicago-land. Recently, the office ordered a new x-ray machine to replace their old and outdated equipment. My mom was very eager to use the new equipment, and was very happy to have an upgrade. However, after a...
Read More -
Requirements | Argentina | Brazil | International | China | Labeling | Registrations | Venezuela | Mexico
Small Label, Big Impact
on 23 January 2013 | By Ryan Kasun
There have been a bunch of countries recently that have changed their regulations to require a native language registration label on medical devices. These labels must call out a specific set of information including manufacturing location and address, and the Registration...
Read More -
Some Things Never Change....
on 23 January 2013 | By Ryan Kasun
There are some things in life that never change. So, if they never change, let’s learn the rules. A teacher once told me “once you know the rules, embrace them.”
Read More -
Medical Devices and Fantasy Football, More in Common Than You Think
on 21 January 2013 | By Ryan Kasun
I constantly find myself trying to draw parallels between my work life and personal life in almost everything I do and while I work in Quality and Regulatory for medical device manufacturers, I enjoy spending downtime playing fantasy football. Being that this weekend was the...
Read More -
Adverse Events | Hip Replacement | Surgery | Safety Communication | Fda | Medical Device | Implantables
Hitting Home
on 18 January 2013 | By Ryan Kasun
One of the more personal aspects of why I work in regulatory and quality is because of my dad. As a youth, he experienced a hip injury. Combine that with a condition (don't ask - I never remember its fancy medical name) that causes weak capillaries, and that resulted in a...
Read More -
Identification... Unique Device Identification.
on 16 January 2013 | By Ryan Kasun
The other night, while I was drinking a refreshing beverage (neither shaken, nor stirred) and completely engulfed in an iconic (British) action-espionage movie, I began thinking of fingerprints (thanks to the movie's protagonist using spiffy spy gear to trick the baddies!) and...
Read More -
Preparing For Fda Visit | Regulatory Harmonization | Remediation | Australia | Fda | New Zealand | Medical Device | Regulatory News | White Paper
Regulatory Intel - Jan. 14, 2013 Quick-Hits
on 14 January 2013 | By Ryan Kasun
A few quick notes I wanted to post while clearing my inbox of my "informative reading" emails from last week:
Read More -
Wellness Products | Watch Sensors | Mobile Medical Applications | Fda | Medical Device | Wearable Tech
Mobile Medical Applications - Healthcare at CES
on 14 January 2013 | By Ryan Kasun
So I wasn't kidding last week about wearable tech, which made the biggest splash at CES 2013, an annual consumer electronics expo. Coverage thanks to CNET.com.
Read More -
Rta | Submission | Fda | Medical Device | 510(K) Submission | 510(K) Review | Guidance Document | Refuse To Accept | 510(K)
New "Refuse to Accept" Guidance Document for 510(k)s
on 14 January 2013 | By Ryan Kasun
There has been much controversy about the new RTA FDA Guidance document titled "Refuse to Accept Policy for 510(k)s". The draft of this guidance was issued for review in August of 2012 and was recently released at the end of that year. However companies are still talking about...
Read More -
Regions | Orthopedic Capital Of The World | Medical Devices | Biomedical | Pittsburgh | Catheter Valley | Ohio | Biomedical Industry
Location, Location, Location
on 10 January 2013 | By Ryan Kasun
January is usually that time of year where I start day dreaming about a vacation somewhere warm and sunny. California is always on the list. California has a lot more to offer than just its beautiful beaches and amazing sights. A recent article on the biomedical industry had...
Read More -
Be Smart: Look Smart
on 9 January 2013 | By Ryan Kasun
I wanted to write about an interesting event that a group of us at RQS had this weekend. One of our employees, Kathy Callan, is a professionally trained Fashion Consultant. Kathy is one of those people that always looks put together. She is walking evidence of her talent and...
Read More -
The Promise
on 8 January 2013 | By Ryan Kasun
Training is an integral part of what we do at RQS, whether it’s one on one or in a more formal setting. Recently, as I talked with staff members at one organization, I realized that we needed to do a basic class on documents that make up the Quality Management System. As process...
Read More -
Quality Lessons of a New Parent
on 7 January 2013 | By Ryan Kasun
2012 brought many new challenges to my life. Some dealt with changes in my professional career, some dealt with personal losses of friends and family, but none were greater than becoming a parent for the first time. Being in Quality I'm accustomed to having some semblance of...
Read More -
Mobile Medical Applications - Only a Bandage?
on 4 January 2013 | By Ryan Kasun
Often enough, I'll hear or say, "It's just a bandage" (to avoid the popular brand-name bandage) in response to a solution that won't truly fix a problem or is just a temporary measure or bridge to the better solution. I had a thought today that it might apply to our current use...
Read More -
Ecopy | Federal Register | Rta | Proposed Rules | Mdufa Iii | Food And Drug Administration Safety And Innovation | Medical Device Excise Tax | Medical Device Industry | Mdufa | Medical Device Establishment Registration And List | Comparative Effectiveness Research | 2013 | Cer | Public Comments | Fdasia | Ecopy Program For Medical Device Submissions | Petition
New Year, New Resolutions
on 3 January 2013 | By Ryan Kasun
Happy New Year - I hope that all of our blog readers had a wonderful holiday and brought in 2013 with warm and welcoming arms!
Read More -
A Fresh Start
on 1 January 2013 | By Ryan Kasun
Every New Year, many of us take stock of our lives and make resolutions to live differently. We see January 1st as an opportunity for a fresh start. Whether it’s the chance to renew a friendship, correct a mistake, or take an opportunity that we may have previously missed,...
Read More -
Regulatory Intel - Alerts, Updates, Newsletters
on 28 December 2012 | By Ryan Kasun
Quite appropriate for the season, I found a little "gift" in my documents folder - a list of Regulatory Intelligence Sources! If I'm remembering correctly, these links may have come from the RAPS Online University program. This week's post: Alerts, Updates, and Newsletter...
Read More -
Unwanted Gifts
on 27 December 2012 | By Ryan Kasun
Have you ever received a gift that you didn’t really want? Was it an ugly sweater from Great Aunt Margie, or a holiday pillow that didn’t quite match with your décor? People always say, it’s the thought that counts… but if that was the case, gift receipts would not be so...
Read More -
Mobile Medical Applications - Progress?
on 21 December 2012 | By Ryan Kasun
I'll admit, I'm a sucker for the "Best of " everything for whatever reason. Why do I need to watch a mash-up of Gangam Style, Walk Off the Earth, and "Call Me Maybe"? The answer is "I don't", but I love it all. Sports stories, worst media sensationalist stories (such as today's...
Read More -

Heart to Heart
on 20 December 2012 | By Ryan Kasun
Seven years ago today, I waited by the phone. Waiting for the news that my niece had been born. This wait was much different for me than previous births. This was a nervous and unsure wait.
Read More -
Mistake, I Think Not!
on 20 December 2012 | By Ryan Kasun
As a native Clevelander and lifelong (i.e. 47 years) Northeast Ohio resident, I have heard my fair share of “Mistake on the Lake (Erie)” jokes. Yes, it’s true the Cuyahoga River caught fire in 1969, the city has suffered from loss of jobs due to de-industrialization, there have...
Read More -
Biodegradable Circuits
on 19 December 2012 | By Ryan Kasun
When I first heard the words “biodegradable” and “electronic devices” together, especially in the context of nanotechnology and the medical field, the idea smacked ever so slightly of science fiction. You know, the sort of advancement that belongs in an era of teleportation and...
Read More -
Astronaut Icecream and Medical Devices
on 18 December 2012 | By Ryan Kasun
In recent years, my father has picked up a new hobby of star-gazing. What started out, as I believe, as an excuse to get out of the house and away from the mother-in-law, has slowly turned into a full-fledged astronomical geekfest (no pun intended). So, for Christmas this year,...
Read More -
Mdd | Eu | Label | Regulatory Affairs | Rqs | Labeling | Ifu | Fda | Health Canada | Medical Device | User Guide | Instructions For Use | Manual
Labels impact the device
on 17 December 2012 | By Ryan Kasun
During this holiday season, when presents are being packaged and shipped at high volumes, I thought it would be appropriate to talk about the importance of labeling.
Read More -
Call me a (Juris) Doctor!?
on 17 December 2012 | By Ryan Kasun
I was going to write about China Labeling this week, but I didn't feel like it!
Read More -
Biocompatibility | Mobile Medical Applications | Precedence | Fda | Substantial Equivalence | Regulatory | 510(K) | Quality
Setting Precedence
on 13 December 2012 | By Ryan Kasun
I sat down tonight, as I do every night that I write a blog post, and I started reading the prior posts from my colleagues. I had every intention of writing on a different topic tonight, but something struck a chord with me when I looked through the blogs. They all had something...
Read More -
Biocompatibility | Mdd | Eu | Medical Devices | 10993-1 | Fda | Health Canada | Medical Device | Medical Device Directive | 10993
Biocompatibility - Simple, Right?
on 12 December 2012 | By Ryan Kasun
I’ve been working in regulatory for a company in which biocompatibility of device materials has been pretty straightforward and understood by all for many years. Annex A in ISO 10993-1 and FDA General Program Memorandum - #G95-1 is all you need. Check the chart for our device...
Read More -
In Your Face
on 11 December 2012 | By Ryan Kasun
My teenager works at a local eatery – I thoroughly believe the saying, “A good teenager is a tired teenager”. Besides, there is a lot to be learned from being in the work force.
Read More -
What is the Medical Device Excise ?
on 10 December 2012 | By Ryan Kasun
Let me start my blog off by stating that I personally love tracking politics. Presidential election years are my favorite, with all the debates, mudslinging, scandals and whatnot that comes with the elections. It's pretty much like watching your favorite reality tv show....
Read More -
Medical Device Innovation Consortium | Raps | Off-label Promotion | Webinar | Mdd | Eu | Mhealth | Medical Devices | Mobile Medical Applications | Training | Fda | Scanadu | Pharma | Medical Device Directive | Mobile Medical Apps | Us Vs. Caronia | Caronia | United States Vs. Caronia | Qualcomm | X Prize
Quick Hits - December 11, 2012
on 10 December 2012 | By Ryan Kasun
In a bit of a change for today's post, I'm going to highlight some of the big news items that came through my inbox last week:
Read More -
 Mhealth | Mobile Medical Applications | Submission | Fda | Devices | Medical Device | Apps | Alivecor | Software | Predicate | Mobile Medical Apps | Ekg | 510(K)
Mhealth | Mobile Medical Applications | Submission | Fda | Devices | Medical Device | Apps | Alivecor | Software | Predicate | Mobile Medical Apps | Ekg | 510(K)Mobile Medical Applications - Community Bull-Dozers
on 6 December 2012 | By Ryan Kasun
Well how about that - software developers are making it easy on me to write this series with yet another 510(k) clearance announced this week. The latest in cleared mobile health devices? The AliveCor mobile heart monitor.
Read More -
New Heights in Prosthetics
on 5 December 2012 | By Ryan Kasun
About a week ago I read an article in Mechanical Engineering magazine that I found inspirational from several perspectives. Design-2-Part online magazine also covers the same story in this article.
Read More -
United States v. Caronia
on 4 December 2012 | By Ryan Kasun
Yesterday, the United States Court of Appeals for the Second Circuit (based in NYC) made a 2-to-1 decision on United States v. Caronia (just one of many criminal cases pertaining to off-label use). This case even contained a supplemental briefing that had the litigants brief the...
Read More -
Women in Bio Success!
on 3 December 2012 | By Ryan Kasun
This past Thursday (11/29) several RQS employees were fortunate enough to attend the kick-off event for the Women in Bio Pittsburgh chapter which featured keynote speaker Alexandra Drane. Alexandra is the founder and Chief Visionary Officer at Eliza Corporation. The event was...
Read More -
Iphone | Mobile Medical Application | Mhealth | X Prize Foundation | Tricorder | Mobile Medical Applications | Android | Medical Apps | Fda | Medical Device | Scanadu | Star Trek | Apps | Blackberry | 510(K) | Qualcomm | Ios
Mobile Medical Applications - Beam Me Up, Scanadu
on 30 November 2012 | By Ryan Kasun
In a fantastic case of science-fiction-meets-reality, Qualcomm created the X Prize Foundation to challenge developers and innovators to create a Star Trek Tricorder. The prize? $10M!
Read More -
Deals and Steals
on 29 November 2012 | By Ryan Kasun
Thanksgiving night I was standing outside in the cold in a line that wrapped halfway around Target. My motivation for this spontaneous Black Friday door-buster shopping was peer pressure, but I was also excited to take advantage of the great prices. Even though I wasn't in the...
Read More -
Quality Systems | Production Quality | Design Quality | China | Fda | Medical Device | Regulatory | Quality
International Spotlight - China
on 29 November 2012 | By Ryan Kasun
When people ask my opinion of the most difficult country to register medical devices internationally, I would have to say it is China. China is considered to be an “emerging market”- it is getting more and more lucrative to register there as the population gets larger and...
Read More -
No Time for Common Sense
on 27 November 2012 | By Ryan Kasun
Last week, I had a computer issue...not a big issue, more like an annoyance. Nevertheless, I dutifully called our IT support and after a couple of hours of investigation, we determined that the problem could be solved by one of two solutions; change the battery or change the...
Read More -
Can't we just all get along?
on 27 November 2012 | By Ryan Kasun
Why is it that anytime a project is underway that it seems as if quality is an afterthought? Why are Regulatory and Quality (R&Q) Engineers typically considered to only support the deliverables at the end of a projects launch? How can this be when they are working our tails off...
Read More -
Balancing Act
on 21 November 2012 | By Ryan Kasun
Early in product development, it’s easy to be overly optimistic about designing a user-friendly device. The risk management process tends to be a reality check of sorts, often resulting in a tradeoff of usability for safety with the intent of mitigating risks. Design changes are...
Read More -
Doctor Mario to Surgery, please… Doctor Mario to surgery
on 20 November 2012 | By Ryan Kasun
When you think of video gamers or computer gamers, what typically comes to mind? Out of the many images, word associations, and stereotypes that may have just gone through your head, I am confident that the words, “future robotic surgeon” were not included.
Read More -
Truth Behind Facts
on 20 November 2012 | By Ryan Kasun
My focus lately has wildly been on healthy eating, various diets and finding the true source of where our food comes from; behind the labeling/packaging, behind the marketing....what am I really eating? Prior to this new obsession food has always just been...food. Something I...
Read More -
Women in Bio - November 29th Kick-off Event
on 20 November 2012 | By Ryan Kasun
On November 29th the Women in Bio (WIB) Pittsburgh chapter will be having its official launch event with guest speaker Alexandra Drane. This will be an interesting informational and networking event that a number of RQS employees are planning to attend. It is encouraging to see...
Read More -
Mobile Medical Applications - Call Security!
on 16 November 2012 | By Ryan Kasun
In an odd turn of coincidence, I have been involved in more discussions regarding mobile security this week than I have in the life of the RQS blog. I took it as a sign that I should write about it.
Read More -
Regulatory Intel - Government Databases
on 16 November 2012 | By Ryan Kasun
One challenge that many regulatory professionals face is finding applicable and relevant data to conduct their day-to-day activities. Regulatory intelligence is important in the constantly evolving healthcare industry for activities such as submissions, pre- and post-market risk...
Read More -
Exploding Bombs?
on 15 November 2012 | By Ryan Kasun
I have to admit, school doesn't teach you everything. During my first week at RQS, I was working with a client, and we were discussing labeling. She said I needed to get the bomb. Then, if I explode the bomb, everything will be good, and I'll have what I need. I left her office...
Read More -
Set Phasers on Stun
on 14 November 2012 | By Ryan Kasun
Several years ago a good friend who knows my interest in human factors and engineering psychology gave me a book called Set Phasers on Stun and Other True Tales of Design, Technology, and Human Error, by Steven Casey. I started to read it again recently and wanted to share it...
Read More -
Rethinking Innovation
on 12 November 2012 | By Ryan Kasun
One of my favorite topics about the medical device industry and technology in general is innovation. Rebecca's post last week discussed the Medical Innovation Summit - "Innovative to the Bone" hosted by the Cleveland Clinic. Although I was unable to attend the event, I did find...
Read More -
Magnetic Resonance Imaging | Medical Imaging | Mobile Medical Applications | Mma | Mri | Medical Device | Computed Tomography | Ipad Medical Apps | Picture Archiving Communication System | Teleradiology | Mobile Medical Apps | Aycan Mobile | Pacs
Mobile Medical Applications - Aycan 510(k)
on 9 November 2012 | By Ryan Kasun
To continue our product highlights within mobile health, we turn to Aycan's "Aycan Mobile" product which received clearance on September 12, 2012. A picture of its use on an iPad is shown on their website.
Read More -
When the Lights Go Down in the City
on 9 November 2012 | By Ryan Kasun
Just the other day, I left work with high ambitions of going to the gym. However, when I walked out of the office, it was completely dark outside; it felt like the middle of the night. I instantly started second guessing my decision, I mean, it had to be almost bed time, right?...
Read More -
Stay out of Frying Pans and Fires!
on 9 November 2012 | By Ryan Kasun
Quick note today as a training seminar came through my inbox.
Read More -
Innovative To The Bone | Rqs | Top 10 Medical Innovations For 2013 | Medical Innovation Summit | 2012 Medical Innovation Summit | Cleveland Clinic | Top 10 Innovations
Innovative to the Bone
on 9 November 2012 | By Ryan Kasun
Last week, Regulatory and Quality Solutions participated in Cleveland Clinic’s 10th Annual Medical Innovation Summit – “Innovative to the Bone”! Each year, the Summit highlights different areas of specialization, with this year focusing on orthopaedics. It was our first year...
Read More -
Lessons of a High School Speech Geek
on 7 November 2012 | By Ryan Kasun
When I was in high school, I was a speech geek. Every Saturday during the Forensics season I got up before the crack of dawn, met up with my speech and debate team, and drove to a different local school to compete in a public speaking tournament. The category I competed in...
Read More -
Searching for Medical Device Info
on 5 November 2012 | By Ryan Kasun
As a recently new professional in the medical device industry, I try to find any tips or tools I can use to gain more knowledge about the industry as a whole. Increasing my general knowledge of the field can only help me when I interact with other professionals. When thinking of...
Read More -
 Iphone | Touchscreen | Mobile Medical Applications | Usability | Buttons | Medicaldevices | Android | Medical Device | Medicaldevice
Iphone | Touchscreen | Mobile Medical Applications | Usability | Buttons | Medicaldevices | Android | Medical Device | MedicaldeviceMobile Medical Applications - Difficult Usability?
on 5 November 2012 | By Ryan Kasun
The topic of this week's post was a bit more challenging than normal. Ever since my inadvertent "reboot" of the Everyday Usability series, my brain was stuck in that frame of mind. Even today I struggled to break my mind from it.
Read More -

Training and a couple jokes
on 2 November 2012 | By Ryan Kasun
Trick-or-treating in my neighborhood was moved to Saturday this year because of Hurricane Sandy...therefore I needed a few Halloween jokes to keep me going until this weekend. Here you go:
Read More -
Left Wing, Right Wing, or Somewhere in Between
on 1 November 2012 | By Ryan Kasun
In the spirit of November’s highly anticipated Presidential Election, an important and multi-faceted question was raised during a recent RQS presentation. “How will the results of this year’s 2012 Presidential Election affect the medical device industry?”
Read More -
Need a Hand?
on 31 October 2012 | By Ryan Kasun
Have you ever felt like you were trying to do too many things at once and could really use a third hand? Like trying to answer the phone while carrying the groceries in (as the bags are breaking) while keeping the dog from escaping…. As our lives get increasingly busier, our...
Read More -
Conducting a Literature Review
on 30 October 2012 | By Ryan Kasun
One issue that came up last week with a client was how to conduct a literature review and how important literature reviews were. I have experienced in some cases, companies that have a lot of man power behind the reviews but these companies do not have solid criteria for...
Read More -
Mdd | Eu | Europe | Medicaldevices | Medical Device | European Union | Medical Device Directives | Ivd | Medicaldevice | Active Implantable Medical Device
Overview of New Medical Device Regulations in Europe
on 26 October 2012 | By Ryan Kasun
This week my inbox was flooded by notifications of a great blog post from the Emergo group, outlining the changes set to occur to the medical device regulations in Europe. Changes such as:
Read More -
 Everyday Usability | Landfill | Usability | Medicaldevices | Starbucks | Medical Device | Trash | Green Movement | Medicaldevice | Recycling
Everyday Usability | Landfill | Usability | Medicaldevices | Starbucks | Medical Device | Trash | Green Movement | Medicaldevice | RecyclingBlast from the Past - Everyday Usability
on 26 October 2012 | By Ryan Kasun
I couldn't resist the urge to post another picture in the Everyday Usability series after seeing this:
Read More -
Mhealth | Idiom | Mobile Medical Applications | Mma | Medicaldevices | Medical Device | Apps | Titanic | Medicaldevice
Mobile Medical Applications: Titanic Chairs
on 26 October 2012 | By Ryan Kasun
I've often believed that the number of idioms and quirky phrases describing a particular characteristic, event, or circumstance is mostly equivalent to its frequency in everyday life.
Read More -
October brings awareness to women’s health issues
on 25 October 2012 | By Ryan Kasun
October is a very important month for women: It’s National Breast Cancer Awareness Month. There are many things to be proud of for how far we have come with treating breast cancer. The Susan G. Komen Foundation reports that the 5-year survival rate for female breast cancer...
Read More -
User Research Methods
on 25 October 2012 | By Ryan Kasun
So you’re designing a new medical device and you want to gain a better understanding of how it will fit into your intended users’ workflow. Where do you start? Some common tools for user research that are particularly useful during early stages of design are contextual inquiry,...
Read More -
Great Lakes Venture Fair
on 24 October 2012 | By Ryan Kasun
Last week, Regulatory and Quality Solutions, attended the first Great Lakes Venture Fair (GLVF). The fast-paced day-and-a-half event (Wednesday, October 17, 2012 and Thursday, October 18, 2012) was held in downtown Cleveland, OH and featured some of the highest potential...
Read More -
Keeping up with Standards
on 23 October 2012 | By Ryan Kasun
With recent clients I have been dealing more and more with new standards coming out or revisions being made to standards. This has got me thinking a lot about how to keep up with current standards. What is the best way of being notified of new releases of standards as well as...
Read More -
Mhealth | Medical Devices | Mobile Medical Applications | Windows 8 | Mma | Medicaldevices | Android | Medical Device | Mobile Medical Apps | Medicaldevice | Ios
Mobile Medical Applications: Whale Cupcake
on 19 October 2012 | By Ryan Kasun
A good friend of mine in high-school was trying to understand the definition of a "hyperbole". Now, if Google would have been sophisticated enough at the time to include the "define:" feature, it would have returned the following for hyperbole: Exaggerated statements or claims...
Read More -
Blockbuster is no more - A discussion on disruptive technology
on 19 October 2012 | By Ryan Kasun
Every time I go home to visit my parents, one thing that comes up more often than not, is the fact that our local Blockbuster closed. It’s been about two years now, and they still can’t get over it. Me? I’m happy to rent from a RedBox, or even better, kick my feet back on the...
Read More -
-
Nano Technology | Frontiers In Optics | Spider Silk | Optics | Nano | Medical Device Industry | Omenetto | Osa | Biomedical | Fio | Huby | Microchip | Optical Society | Implantables | Spider-man
Spider silk isn't just for Spider-Man
on 17 October 2012 | By Ryan Kasun
The vast majority of us are familiar with Stan Lee and Steve Ditko’s Marvel comic book series, "The Amazing Spider-Man", where he would go web-slinging from here to there, in order to catch the baddies (of course, there is a lot more to this story than just that… but...
Read More -
Medical Devices in Different Markets
on 16 October 2012 | By Ryan Kasun
I recently took a two week vacation to South Africa traveling to Cape Town, Prince Albert and various parts of the northern Western Cape. It was an absolutely amazing trip with many memories of time spent with my family. Traveling to a foreign country has shown me a lot of new...
Read More -
Medical Device Tax: Funding Healthcare for Millions
on 12 October 2012 | By Ryan Kasun
In the second of the two part series, we'll review support of the 2.3% tax of the medical device industry to go into effect on January 1, 2013. Earlier this week, I outlined why the industry sees this tax as an enormous burden and described actions already being taken to off-set...
Read More -
Devices Regulations in Europe
on 11 October 2012 | By Ryan Kasun
On Tuesday, I attended a Pittsburgh Technology Council event downtown titled “Medical Devices and the European Union: What does the Path of Entry Look Like?” which was sponsored by RQS! (We gave out stress balls in the shape of a pill!) I’ve posted a few times before regarding...
Read More -
Now that’s some real horse power
on 11 October 2012 | By Ryan Kasun
Our neighbors located up the street from our Cleveland, OH location, at the Cleveland Clinic, have recently helped develop horse power into a whole new meaning. A Cleveland Clinic researcher found that one reason horses are more efficient at walking and running than humans, is...
Read More -
Moderating Usability Testing
on 10 October 2012 | By Ryan Kasun
Since I’m in the middle of several usability testing projects right now, I thought this would be a great time to discuss some things to remember when moderating usability testing activities:
Read More -
Medical Device Tax: Windfall or Pitfall?
on 9 October 2012 | By Ryan Kasun
Thanks to Sherri's earlier post, we've begun to explore the landscape of the medical devices industry in reaction to the 2.3% tax on revenue for medical device companies as a result of the Affordable Care Act. The tax is being implemented in part to fund the estimated 30 million...
Read More -
Creating Consistency
on 8 October 2012 | By Ryan Kasun
One major variable med tech companies are faced with today is the unknown of FDA reviewers. It seems like what works for one company doesn’t necessarily work for the rest. And why is this so?
Read More -
Navigating Substantial Equivalence
on 5 October 2012 | By Ryan Kasun
Part of the mission of the CDRH is to ensure medical devices are safe and effective. Based on the risk of the device and the agency’s familiarity with the device type, different levels of evidence are required to convince the FDA that a device is safe and effective.
Read More -
Occupational Safety And Health | Osha Guidelines | Mdd | Eu | Medical Devices | Intuitive Responses | Design Considerations | Usability | Interaction | Occupational Safety And Health Administration Osha | Environments | Fda | Health Canada | Medical Device | Occupational Safety And Health Administration | 510(K) | Environmental Conditions
Mobile Medical Applications: Mobile Medical Device Design
on 3 October 2012 | By Ryan Kasun
Many medical devices are meant to be used while in mobile situations. These devices could be wearable, carried in a user’s pocket or by hand, pushed like a cart, etc. Users of mobile medical devices expect them to be safe, reliable, durable, and easy to use while in motion or...
Read More -
More changes heading our way?
on 3 October 2012 | By Ryan Kasun
After a recent statement by the EU health commissioner, it appears that FDASIA's influence may have gone abroad. As the medical device industry gears up and is adapting to the changes that FDASIA brings, it may also have to prepare itself for even more changes heading our way.
Read More -
RQS on YOUR Airwaves
on 1 October 2012 | By Ryan Kasun
Another thanks must go out to the Pittsburgh Technology Council for hosting RQS on their TechVibe Radio program. The PTC's CEO Audrey Russo and Director Visibility Initiatives Jonathan Kersting interviewed President Maria Fagan, Vice President Lisa Casavant, and Vice President...
Read More -
Medical Devices | Healthcare Innovation | Mobile Health | Medical Claims | Health It | Small Business | Fda | Algorithm | Himta | Mobile App Regulation | Fdasia | Mobile Apps
Mobile Medical Applications: Innovation vs. Regulation
on 1 October 2012 | By Ryan Kasun
Mobile apps are absolutely exploding on the marketplace. It seems like so many business entrepreneurs are focusing on creating new apps that fit various needs such as games, music, organizational purposes and the list goes on. I recently read an article that discussed creating a...
Read More -
Medical Device Design for Cross-Cultural Use
on 26 September 2012 | By Ryan Kasun
As if it isn’t challenging enough to design a medical device that can be used safely and effectively by a select group of individuals in a specific environment, the complexity increases when designing for international use by operators with diverse cultural backgrounds....
Read More -
 Iphone | Mhealth | Diabetes | Glucose Measurements | Mobile Health | Mobile Medical Applications | Electronic Log Book | Regulatory Department | Android | Fda | Medical Device | Regulatory News | Apps | Design Engineers | Device Manufacturers | Glucose Monitor | Medical Application | Fda Clearance
Iphone | Mhealth | Diabetes | Glucose Measurements | Mobile Health | Mobile Medical Applications | Electronic Log Book | Regulatory Department | Android | Fda | Medical Device | Regulatory News | Apps | Design Engineers | Device Manufacturers | Glucose Monitor | Medical Application | Fda ClearanceMobile Medical Applications: Falling Star?
on 26 September 2012 | By Ryan Kasun
I think it's high time we start profiling a few applications that have been approved for use by the FDA, discuss their functionality, and potentially profile the impending issues they may encounter.
Read More -
Networking Success!
on 24 September 2012 | By Ryan Kasun
The Networking with Technology event hosted by the Pittsburgh Technology Council and Pittsburgh Social Exchange last Wednesday on September 19th was a success!
Read More -
Unexpected Benefits
on 20 September 2012 | By Ryan Kasun
Medical Device Companies are required to comply with various regulations, based on where products are marketed. As start-up businesses or established companies enter the medical device arena, sometimes quality systems and regulations are viewed as a painful part of doing...
Read More -
Setting Realistic Expectations of Medical Device Users
on 19 September 2012 | By Ryan Kasun
During recent usability testing of an updated medical device, I was reminded how important it is to set realistic expectations of medical device users when designing a product. Since the end users are the experts at what they do, it’s easy to think of them as having super-human...
Read More -
Networking with Technology
on 18 September 2012 | By Ryan Kasun
Several RQS employees will be attending the Networking with Technology event this Wednesday hosted by the Pittsburgh Technology Council and Pittsburgh Social Exchange. This event is taking place on Wednesday the 19th from 6:00-8:00 PM at the Rivers Club. The event will provide...
Read More -
Smartphones | Iphone 5 | Iphone | Medical Devices | Google | Application Development | Dual Core | Lumia | Mobile Health | Mobile Medical Applications | Nexus | Health Applications | Fda | Windows Millenium | Technical Specifications | App Development | Qualcomm
Mobile Medical Applications: Five or One Galaxies in the Lumia Nexus?
-
Standing Out vs. Fitting In
on 13 September 2012 | By Ryan Kasun
Innovators are constantly being pulled in different directions on whether they should develop a product that ‘Stands Out’ or one that ‘Fits In’. It’s like middle school all over again! I’ll go through some of the challenges with choosing to develop a new state of the art device...
Read More -
Cleveland, We Have a Problem
on 12 September 2012 | By Ryan Kasun
Since this is my first contribution, let me give you the intro - my name is Mike Andreas and I am a Senior Quality Engineer with RQS. My background, aside from 5 years in the medical imaging industry, includes mechanical engineering, configuration management, and a lot of pizza....
Read More -
Healthcare going mobile
on 11 September 2012 | By Ryan Kasun
Hospital patients (and hospital staff) no longer have to grow weary of the constant due diligence of power cord avoidance (in fear of tripping on or becoming tangled in cords from the patient monitoring devices). Now, thanks to wireless medical devices, such as portable patient...
Read More -
Creating Growth and Improving Performance
on 10 September 2012 | By Ryan Kasun
I recently read an article that focused on describing what it takes for a company to create growth and improve its performance. So how can companies do more with less? This article describes several areas a company can focus on in order to become more successful in their...
Read More -
RQS on the Airwaves!
on 7 September 2012 | By Ryan Kasun
Thanks to the Pittsburgh Technology Council and their TechVibe Radio program, Regulatory and Quality Solutions will be on the radio this Saturday at noon. Hosted by the PTC's CEO Audrey Russo and Director Visibility Initiatives Jonathan Kersting, the radio program will provide a...
Read More -
Death and Taxes
on 6 September 2012 | By Ryan Kasun
We’ve heard it before: “There are two things certain in life: death and taxes.”
Read More -
Success Rates for Usability Objectives
on 5 September 2012 | By Ryan Kasun
Continuing our discussion about usability objectives from last Wednesday, a related question that comes up frequently is – What acceptance criteria should I use for my usability objectives? or Is it acceptable to have a usability objective of less than 100% success?
Read More -
Nanomaterials | Medical Device Industry | Medical Device | Nanotechnology | Fdasia | Med-tech Industry
Nano-what? Nanotechnology.
on 5 September 2012 | By Ryan Kasun
Over the past 50 years, the advancement of technology has been beyond incredible. Although, we are not zipping around in flying cars, fearing the dreaded encounter with a hoverboard gang, or living amongst cyborgs, we are witnessing things never before thought possible. Such...
Read More -
Becoming a Successful Consultant
on 4 September 2012 | By Ryan Kasun
Since I am a recent graduate and new to the work force, I wanted to discuss what exactly it takes to become a successful regulatory affairs consultant in the life science industry. Biospace wrote an article on this subject describing the five steps to becoming a successful...
Read More -
PTC Breakfast Briefing on EU Market
on 31 August 2012 | By Ryan Kasun
Pittsburgh Technology Council (PTC) is hosting a Breakfast Briefing on October 9th, 2012 to discuss important aspects on gaining approval and releasing medical device product in the European Union. The briefing will include a keynote by Paul Brooks of the British Standards...
Read More -
FDA Releases Mission, Vision, and Shared Values
on 31 August 2012 | By Ryan Kasun
The core of strong organizations can be found in its mission and its vision. The Center for Devices and Radiological Health (CDRH), the body of the FDA dedicated to medical devices, strives, just like an private company, to be a strong organization. In an attempt to meet this...
Read More -
What's taking so long?
on 30 August 2012 | By Ryan Kasun
How long does it take to get device clearance through the 510(k) pathway? Does your 90 days seem to stretch into 160? What’s taking so long?
Read More -
FDA: Jekyll or Hyde?
on 29 August 2012 | By Ryan Kasun
Black and white. Yes and no. On and off. Absolutes. Extremes. Exactly what our sensationalist media involuntarily expects each of us to believe is absolute truth. And, thankfully, wisdom typically prevails with the ever-present maxim “The truth lies somewhere in between.”
Read More -
Defining Usability Objectives
on 29 August 2012 | By Ryan Kasun
If you ever plan usability testing activities for a medical device, you may find yourself responsible for defining usability objectives. Today’s topic is - What makes a good usability objective?
Read More -
Walking like an Egyptian
on 28 August 2012 | By Ryan Kasun
Recently, the little brother of my close childhood friend and his family experienced a dramatic, life-altering event. While serving in the US Army as a Commanding Officer of the Army Rangers, my friend’s little brother became a double amputee while on tour in Afghanistan.1,2...
Read More -
RQS Honored With Tech 50 Nomination
on 27 August 2012 | By Ryan Kasun
Yearly, the Pittsburgh Technology Council issues awards, the Tech 50 awards, for local technology companies to recognize the success and growth of the technology sector in the Pittsburgh region. RQS is pleased to announce that it has been nominated for a 2012 Tech 50 award in...
Read More -
An Idea Around Innovation
on 27 August 2012 | By Ryan Kasun
Hello and welcome! This is the kickoff blog to RQS’s new daily blogging social media initiative. The purpose of this blog is to provide current, thought-provoking content about quality and regulatory affairs, life sciences and healthcare that impacts the Pittsburgh and Cleveland...
Read More -
RQS Rises to Top of Pittsburgh 100
on 24 August 2012 | By Ryan Kasun
As announced Thursday at a ceremony and reception hosted by Pittsburgh Business Times at the Duquesne Club, Regulatory and Quality Solutions was named the 4th fastest growing company overall in the Pittsburgh region, and 2nd fastest in the Professional Services category.
Read More -
RQS in the News - Cleveland Office Opening
on 5 April 2012 | By Ryan Kasun
Regulatory and Quality Solutions is in the news again, this time featured in the Pittsburgh Business Times. The article describes the Cleveland market and why it is an attractive location for the first branch office of RQS. The article can be found online here.
Read More -
FDA ISO 13485:2003 Pilot Program
on 5 April 2012 | By Ryan Kasun
Effective June 5, 2012, FDA will begin a voluntary pilot program to accept voluntary submissions of external audit reports compiled by one of the founding members of the Global Harmonization Task Force (GHTF). Said audit reports will be reviewed by the FDA to determine whether...
Read More -
Upcoming Webinar - Common Mistakes for Medical Device Startups
on 4 April 2012 | By Ryan Kasun
BSI is hosting a webinar on April 24th, 2012 at 11:00 AM on the 7 common mistakes of medical device startups. I would anticipate that their information stems from their work as a notified body for manufacturers seeking market clearance for Europe (in which case, this information...
Read More -
Titrimetry: A Brief Overview
on 20 January 2012 | By Kevin Rowland, RQM+ Director of R&D
At RQM+ Lab Services, we pride ourselves on our scope of expertise. We rely on a fully grounded comprehension of long-standing and cutting-edge chemical testing methods. This enables us to deliver results via a limitless range of finely tailored analytical workflows. Our PhD...
Read More -
Registration Database | Iphone | Wellness Products | Jawbone | Personal Data | Myfitnesspal | Bodymedia | Workouts | Fda | Mobile Applications | Quick Scan | Fitness And Health | Barcodes | Calorie Burn | 510(K) | Annoyance | Weight Watchers | Mobile Apps | 60 Minutes | Gray Area | Electronic Logs
Mobile Medical Applications: Wellness Apps
on 18 January 2012 | By Ryan Kasun
After a small hiatus, the Mobile Medical Apps blog series is back! Today we'll take a look into wellness and weight-loss applications.
Read More -

The Working Principles of Dynamic Light Scattering
on 4 January 2012 | By Kevin Rowland, Jordi Labs/RQM+ Director of R&D
Dynamic light scattering is a fundamental technique in particle measurement and characterization. It is widely used to illuminate the dimensions of particles in samples as well as their biomechanical and chemical characteristics under distinct conditions.
Read More -
Medical Images | Software Developers | Submissions | Regulatory Effort | Image Viewing Software | Diagnosis | Computer File | Ipad | Regulatory Pathway | Submission | Fda | Medical Device | Image Credit | Barco | Press Release | Calibration | Medical Applications | 510(K) | Princeton | Job | Fda Clearance
Mobile Medical Applications: Visual Calibration
on 2 December 2011 | By Ryan Kasun
Our third installment of the mobile medical apps series finally features a mobile medical app. This app in particular, the MediCal QAWeb Mobile app, is used to calibrate the screen of an iPad for diagnosis of medical images viewed on said iPad (both versions 1 and 2 according to...
Read More -
Iphone | Mdd | Mhealth | Medical Devices | Mobile Health | Mobile Medical Applications | Mma | Fda | Medical Device | Mobile Medical Apps | 510(K)
Mobile Medical Applications: Cyber Security
on 28 November 2011 | By Ryan Kasun
As with all software and technology, security is, and will always remain, a hot-button topic. It is even a point of contention for computer consumers - Mac vs. PC debates often will focus on how Mac's don't "need" virus protection (which is somewhat of a misnomer, but certainly...
Read More -
Iphone | Mobile Medical Application | Mhealth | Mobile Health | Mobile Medical Applications | Mma | Android | Fda | Apps | Blackberry | Smart Phone
Mobile Medical Applications: What's App-ening?
on 22 November 2011 | By Ryan Kasun
It has been predicted by many technology pundits that smartphones will be in everyone's hands within the next few years. Of course there will be different flavors of smartphones to adjust to different consumer ranges, but the way of the numpad flip-phones will be left to the...
Read More -

Everyday Usability 14
on 18 November 2011 | By Ryan Kasun
Well folks, our Everyday Usability photo series has come to an end with this final post. Today's blog post, in line with last week, is the nuance of language. For example, I "googled" the term "Usability" to be inspired by pictures of usability for the post. Alas, I needed the...
Read More -

RQS in Top 25 of Best Places to Work in Western Pennsylvania
on 24 October 2011 | By Ryan Kasun
As announced last Wednesday, October 19th, 2011, Regulatory and Quality Solutions LLC placed 25th of the 78 nominees for "Best Places to Work in Western Pennsylvania in 2011.
Read More -
Everyday Usability 13
on 24 October 2011 | By Ryan Kasun
For our penultimate edition of Everyday Usability, we take a look into what the future of Usability could look like. What better way to improve usability than to remove the physical interface altogether and implementing voice control of medical devices?
Read More -
-

Everyday Usability 12
on 11 October 2011 | By Ryan Kasun
Good morning blog folks! I greet you this morning with a familiar topic from the early part of this usability series: Colors! (or "Colours!" for those of our readers who favour the use of "u" in said words).
Read More -
"Cheap" Microscope Anyone?
on 10 October 2011 | By Ryan Kasun
Biotech's slow coup of the mobile industry never ceases to amaze me. In this latest piece of news, the addition of a simple (and cheap) ball lens to the iPhone camera creates a microscope strong enough to view details at the cellular level. It is being touted as the perfect...
Read More -
Incentive-Based Regulation: Is it possible?
on 5 October 2011 | By Ryan Kasun
As some who know me would tell you, my mind happens to wander at times about a variety of topics. The subject of this mental galavant happened to be the current regulatory landscape in the United States - specifically the FDA's new outlook of regulation and greater enforcement.
Read More -

Everyday Usability 11
on 30 September 2011 | By Ryan Kasun
A great example of usability engineering is right in front of us every day (especially as we become a more technological society): electrical outlets!
Read More -
Improving Device Human Factors Through Focus Groups
on 29 September 2011 | By Ryan Kasun
When designing medical devices it is important to identify use-related hazards as early in development as possible so that mitigations can be developed into user interface requirements, incorporated into the design, and then tested for effectiveness. The recently released draft...
Read More -
Acne Apps Dried Up by FTC
on 26 September 2011 | By Ryan Kasun
In light of the recent drafts of the FDA's regulation and guidance for controlling mobile apps, it is the Federal Trade Commission (FTC) handing down fines to app developers for false claims.
Read More -

Everyday Usability - 10
on 20 September 2011 | By Ryan Kasun
Here in our tenth edition of Everyday Usability, we'll discuss the usability of bathrooms!
Read More -
Disney's Root Cause Analysis?
on 19 September 2011 | By Ryan Kasun
My son was watching Disney's Beauty and the Beast in the car this weekend and I noticed, being the complete nerd that I am, that Belle and the Beast conducted a pretty good root cause analysis:
Read More -
EN 62304 Certification?
on 8 September 2011 | By Ryan Kasun
Last week, I guided a client and their software developer through a notified body audit. Thankfully, this particular developer was well polished (albeit not ISO 13485:2003 certified) resulting in a smooth and quite impressive audit result. The length of the audit was far shorter...
Read More -

Everyday Usability 9
on 7 September 2011 | By Ryan Kasun
Yankees - Redsox. Democrats - Republicans. French - British. Google - Apple. They find themselves in perpetual disagreement; however, I'm sure there is one topic on which no one will disagree:
Read More -
RQS Nominated for 2011 Best Places to Work
on 1 September 2011 | By Ryan Kasun
Regulatory and Quality Solutions, LLC was recently announced as a nominee as one of the "Best Places to Work in Western Pennsylvania". The announcement of the winners will occur later in Fall 2011. The RQS blog will provide updates with any web-based news stories prior to the...
Read More -
FDA Issues Draft Guidance for Clinical Investigations
on 1 September 2011 | By Ryan Kasun
On 8/29/2011, the FDA issued a Draft Guidance recommending changes to monitoring practices in clinical trials. The earlier 1988 Guidance recently was withdrawn and building on the spirit of ICH E6 (1996), the FDA recognizes the desirability of increased monitoring flexibility in...
Read More -

Everyday Usability 8
on 31 August 2011 | By Ryan Kasun
I was discussing quality system documentation with a colleague yesterday when it dawned on me that usability can (and should) apply to quality systems. An example I later identified is shown below:
Read More -
FDA Regulation Goes Mobile
on 30 August 2011 | By Ryan Kasun
Mobile medical applications are revolutionizing the way that healthcare is delivered across the world. In fact, it is estimated that by 2015, 500 million smart phone users worldwide will be using a health care application (app) on their Smartphone or tablet [1]. Mobile medical...
Read More -

Everyday Usability 7
on 22 August 2011 | By Ryan Kasun
There must be something about the simplicity and innocence of children that is particularly inspiring when it comes to usability. Clearly, it is a frequent inspiration for this picture series and this week's post is no exception.
Read More -
PBT Article on RFID Tagging Quotes VP Casavant
on 18 August 2011 | By Ryan Kasun
In the July 29th to August 4th edition of the Pittsburgh Business Times, Kris B. Mamula writes about Ortho-Tag, Inc., a McKees Rocks company that has created an RFID (Radio Frequency Identification) tagging system for implants. The technology will revolutionize the information...
Read More -

Everyday Usability 6
on 15 August 2011 | By Ryan Kasun
Have you ever felt like an idiot for walking straight into a doo
Read More -

IOM Issues Report on the FDA's 510(k) Process
on 10 August 2011 | By Ryan Kasun
The Institute of Medicine (IOM) has released their latest report regarding their review of the 510K process as requested by the FDA. Recall the FDA requested that the IOM review the 510K process on several specific areas to assess the process with regard to protecting the public...
Read More -

Everyday Usability 5
on 9 August 2011 | By Ryan Kasun
Ever attempt to open a carton of milk, but end up splitting the lip of the carton? Of course, the frustrating battle ensues as you struggle to migrate that final sliver to the other side. Clearly, our pain was heard! Companies began to sell their milk with convenient plastic...
Read More -
Scott Wright joins RQS as Principal Regulatory and Quality Advisor
on 8 August 2011 | By Ryan Kasun
Please join me in welcoming team member Scott Wright, Principal Regulatory and Quality Advisor, to RQS!
Read More -
MDDS Regs Change, Provide Insight for Future Changes?
on 2 August 2011 | By Ryan Kasun
The United States Food and Drug Administration (FDA), in an attempt to further the safety of the nation’s medical data systems, issued the final rule for the reclassification of medical device data systems (MDDS) on February 15th, 2011 and went into effect on April 18th, 2011....
Read More -

Everyday Usability 4
on 1 August 2011 | By Ryan Kasun
Prior to becoming a parent, I prepared for many things - diapers, late nights, regurgitation (to be light) - and have experienced them all (plus the many wonderful moments!) in my 13 months as a dad. What I wasn't consciously thinking about were toys - loads and loads of toys...
Read More -

Everyday Usability 3
on 25 July 2011 | By Ryan Kasun
Our VP, suffering from a terrible cold last winter, decided that the best remedy would be the DayQuil/NyQuil dual pack. Perfect, right?! Symptoms dealt with day and night with the added bonus of deep sleep. Alas, the development at Vicks clearly didn't realize that the NyQuil...
Read More -

Everyday Usability 2
on 20 July 2011 | By Ryan Kasun
The Double Wide Grill is an eclectic eatery on Carson Street in Pittsburgh's South Side Flats. Apart from its wonderful culinary offerings, it also offers a fantastic example of usability for this week's post (and, no, not all of these posts will be inspired by restaurants, I...
Read More -
VP Casavant featured in Pittsburgh Business Times
on 17 July 2011 | By Ryan Kasun
Lisa Casavant, Vice President of Regulatory & Quality Solutions, was quoted in a recent article in the Pittsburgh Business Times. In the article, Casavant spoke on the impact of usability on the medical device industry. Usability has become a focus of the FDA and Europe as it is...
Read More -

Picture Series - Everyday Usability 1
on 12 July 2011 | By Ryan Kasun
Today begins Regulatory & Quality Solutions' first picture series titled "Everyday #Usability". The intent of the series is to post images from everyday life that emphasize the importance of usability. There will be examples of both poor and great usability. This week's post?...
Read More -

Pittsburgh Business Times Featured Article
on 6 May 2011 | By Ryan Kasun
Regulatory & Quality Solutions adapts business model to economy
Read More






![FDA Inspections Summit – EU MDR: The Final Push [Panel]](https://www.rqmplus.com/hs-fs/hubfs/Events/2019/RAPS%20Regulatory%20Convergence/IMG_3664-min.jpeg?length=450&name=IMG_3664-min.jpeg)


















![EU MDR / CER Portfolio Planning: The Essential Requirements [Free Webinar]](http://www.rqmplus.com/hs-fs/hubfs/Webinars/RQ/R&Q%20Intelligence%20Series%202017/September%20-%20Portfolio%20Planning/Webinar_Promo_September_Portfolio_Planning_Website-min.png?length=450&name=Webinar_Promo_September_Portfolio_Planning_Website-min.png)






![[Free R&Q Webinar] Cybersecurity for Medical Devices: The regulatory and quality ramifications.](http://www.rqmplus.com/hs-fs/hubfs/Webinars/RQ/R&Q%20Intelligence%20Series%202017/July%20-%20Cybersecurity/Webinar_Promo_July_Cybersecurity_v5_Marilyn_Website-reduced.png?length=450&name=Webinar_Promo_July_Cybersecurity_v5_Marilyn_Website-reduced.png)

![MEDDEV 2.7/1 & CERs: Know the Changes and What to Do [Webinar]](http://www.rqmplus.com/hs-fs/hubfs/Webinars/RQ/R&Q%20Intelligence%20Series%202017/May%20-%20MEDDEV/Webinar_Promo_May_MEDDEV_v2-min.png?length=450&name=Webinar_Promo_May_MEDDEV_v2-min.png)

![EU MDR: Assessing the Impact and Next Steps [Webinar]](http://www.rqmplus.com/hs-fs/hubfs/Webinars/RQ/R&Q%20Intelligence%20Series%202017/April%20-%20EU%20MDR/Webinar_Promo_April_EU_MDR_v2-min.png?length=450&name=Webinar_Promo_April_EU_MDR_v2-min.png)

![MDSAP: What is it? Is it for me? How will I get it done? [Webinar]](http://www.rqmplus.com/hs-fs/hubfs/Webinars/RQ/R&Q%20Intelligence%20Series%202017/March%20-%20MDSAP/Webinar_Promo_March-min.png?length=450&name=Webinar_Promo_March-min.png)


![A Risk-Based Approach to Your QMS Implementation - ISO 13485:2016 and ISO 9001:2015 [Webinar]](http://www.rqmplus.com/hs-fs/hubfs/Headshots/Optimized_for_Web/mark_swanson-min.jpg?length=450&name=mark_swanson-min.jpg)





































































We are passionate about your success. Tell us more about your regulatory and quality needs to learn about how we can help.

GLOBAL BOTTOM CTA INSTRUCTIONS:
To display custom copy instead of global copy in this section, please go to Show Global Content for Bottom CTA? toggle in the "Contents" tab to the left, toggle it off, save, and then REFRESH the page editor, the custom text will then show up and ready to be edited.
Turning the global content back on will be the same process, go to the toggle and toggle it back on, save and refresh!
.svg "white RQM logo")




