
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of current and former FDA reviewers, scientists, engineers and regulatory and quality experts, and adds additional expertise with FDA submissions. The author of this post is a member of this team, which has done significant work with novel and/or high-risk devices focusing on pre-submissions, 510(k)s, IDEs, PMAs, De Novos, Breakthrough Designation Requests and Safer Technology Program Requests.
On occasion, FDA issues exceptions for types of devices that require premarket notification. In their own words, "The FDA believes that the device types identified in the Federal Register are sufficiently well understood and do not present risks that require premarket notification review to provide a reasonable assurance of safety and effectiveness.
This action will decrease regulatory burdens on the medical device industry and will eliminate private costs and expenditures required to comply with Federal regulation."
On April 7 of 2017, FDA published a list of 71 Class I devices that will no longer require premarket clearance. Essentially, FDA has decided that these devices are understood well enough that they don't need to go through the pre-approval process or be found substantially equivalent to an existing device. Class I devices are considered the lowest risk. The example FDA gives of a Class I device is perfect: dental floss.
"FDA classifies medical devices based on the risks associated with the device. Devices are classified into one of three categories—Class I, Class II, and Class III.
Class I devices are deemed to be low risk and are therefore subject to the least regulatory controls. For example, dental floss is classified as Class I device.
Class II devices are higher risk devices than Class I and require greater regulatory controls to provide reasonable assurance of the device’s safety and effectiveness. For example, condoms are classified as Class II devices.
Class III devices are generally the highest risk devices and are therefore subject to the highest level of regulatory control. Class III devices must typically be approved by FDA before they are marketed. For example, replacement heart valves are classified as Class III devices."
I recommend reading the full notification, but here's an excerpt. FDA reviewed Class I devices and made determinations as follows: "...a class I device is exempt from the premarket notification requirements under section 510(k) of the FD&C Act, unless the device is intended for a use which is of substantial importance in preventing impairment of human health or it presents a potential unreasonable risk of illness or injury (hereafter “reserved criteria”). Based on these reserved criteria, FDA has evaluated all class I devices to determine which device types should be exempt from premarket notification requirements. In developing the list of exempt devices, the Agency considered its experience in reviewing premarket notifications for these devices, focusing on the risk inherent with the device and the disease being treated or diagnosed (e.g., devices with rapidly evolving technology or expansions of intended uses). The Agency also considered the history of adverse event reports under the medical device reporting program for these devices, as well as their history of product recalls. Following these considerations, FDA reached the final determination that the devices listed in table 1 do not meet the reserved criteria in that they are not intended for a use that is of substantial importance in preventing impairment of human health or present a potential unreasonable risk of illness or injury."
FDA is always looking for ways to get medical devices to market as safely and efficiently as possible. There tends to be a view that FDA is here to muddy the waters and make things more complicated, but, having been on both sides—currently a regulatory affairs consultant and formerly an FDA reviewer—part of my goal is helping people understand that this is simply not the case. Truly, it's in everyone's best interest to help US citizens get the best and safest healthcare possible! One way FDA does this is by reviewing and updating policies, procedures, and the current status quo. One way industry does this is by following the regulations and general controls to ensure quality devices are entering the market.
If you've attended any of the panels or webinars I've been on where I discuss FDA 101: Tips, Tricks, and Trends, you already know some of the ways that FDA reviews existing policies. Exempting Class I devices from premarket notification is just another way they continue to ensure they're facilitating the process of getting safe and helpful medical devices to market.
Check out the complete table below and additional information on the types of Class I device types exempt from premarket notification.
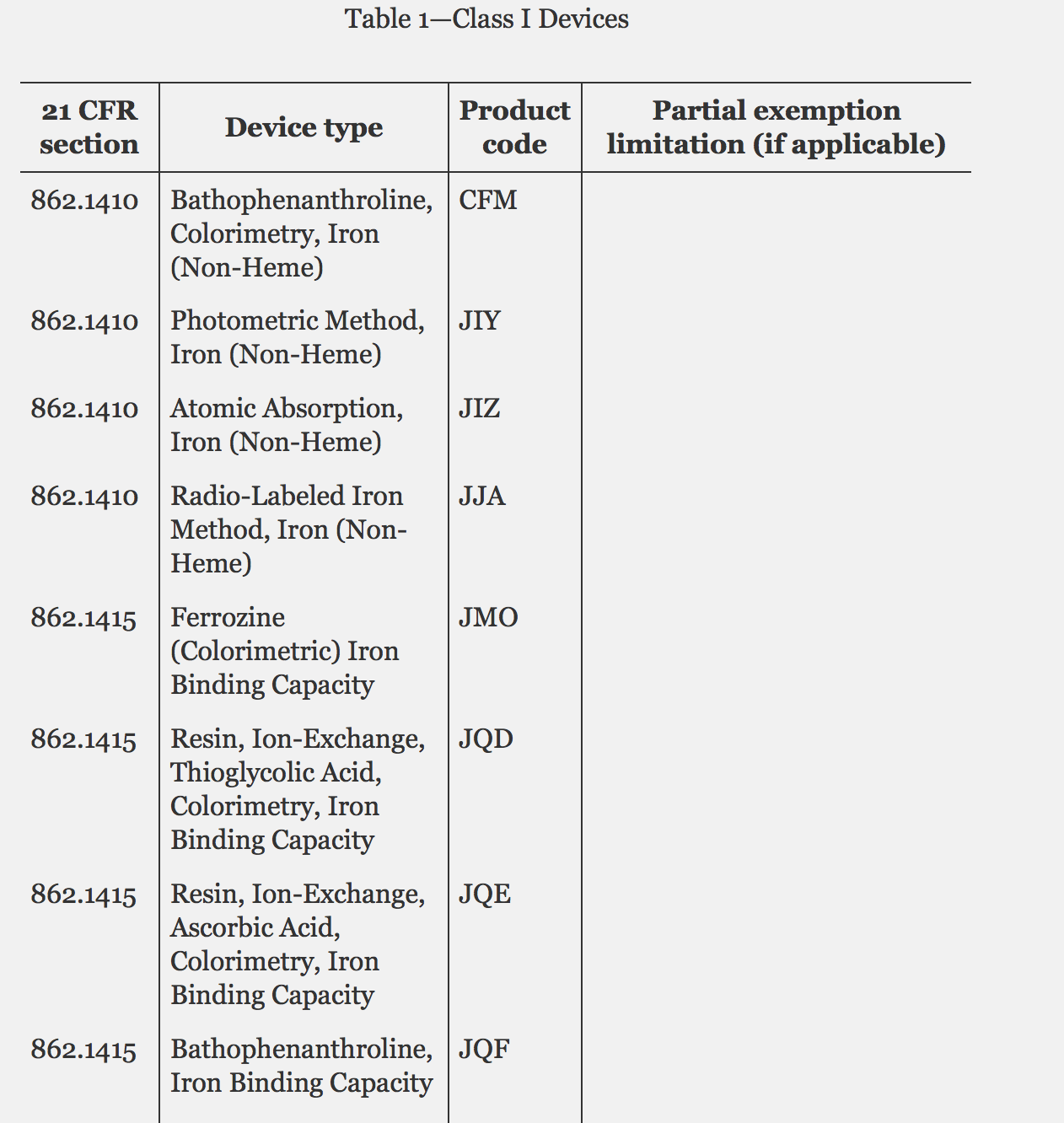
Screen cap from federalregister.gov.
Further Reading (and a podcast!):

.svg "white RQM logo")




