
The EU MDR date of application has come and gone, and most manufacturers are well aware of the new requirements. However, awareness does not necessarily equal effective implementation. One of the main challenges of MDR compliance is creating a strategy that integrates the work of multiple teams, including regulatory, quality, risk management, clinical, and more.
With the benefit of real-world experience across a range of device types and classes, Dr. Jai Kutty and Jon Gimbel discuss these challenges and offer best practices in our webinar, “Best Practices and Processes for Integrating CERs and Post-Market Surveillance Under EU MDR.” This post covers the highlights. For more details and a Q&A, watch the full webinar.
The Risks of Failing to Integrate Processes
Manufacturers are learning that the approach to EU MDR is not just business as usual. New processes and systems must be created to streamline submissions and ensure compliance. Without this internal effort to integrate teams, there can be significant—and costly—consequences.
Efforts may be duplicated.
When teams don’t coordinate their efforts, the result is often inefficiency in creating the documents and a duplication of effort. With multiple deadlines looming, many manufacturers can’t afford to waste resources in this way, not just financially but also in terms of productivity. Take a step back to look at the big picture, and create a process map that clearly defines a workflow with roles and responsibilities to minimize duplication of effort.
Notified bodies might find inconsistencies.
One of the most common—and most avoidable—notified body findings is inconsistency in technical documentation. For example, the intended use and indications for use statement must be the same in the instructions for use, clinical evaluation report, and PMS documents. Furthermore, the benefits and residual risks identified in the clinical evaluation and risk management file must be carried forward into the instructions for use. In many instances, this information is not properly coordinated, which results in notified body findings due to inconsistencies across documentation. The result is rework that could have been avoided if teams had coordinated in the first place.
Deadlines may be missed.
Whether internal or external, repeatedly missing deadlines has repercussions. Missing internal deadlines is tough on the morale of teams because they feel that they are always behind. Coordinating deliverables and making it clear who is responsible for each part of the project is imperative to meeting deadlines. Too often, delays are incurred waiting on updates that could have been avoided with appropriate communication and an alignment on the strategy early in the process.
Missing external deadlines can have major consequences such as a delay in approval, a delay to market, a revoked certificate, and so on. The ripple effect of missing regulatory deadlines should not be underestimated, especially when it comes to protecting the overall health of the business.
Best Practices for Technical Documentation Integration
Fortunately, adopting certain best practices can help you better integrate processes and avoid these potential pitfalls.
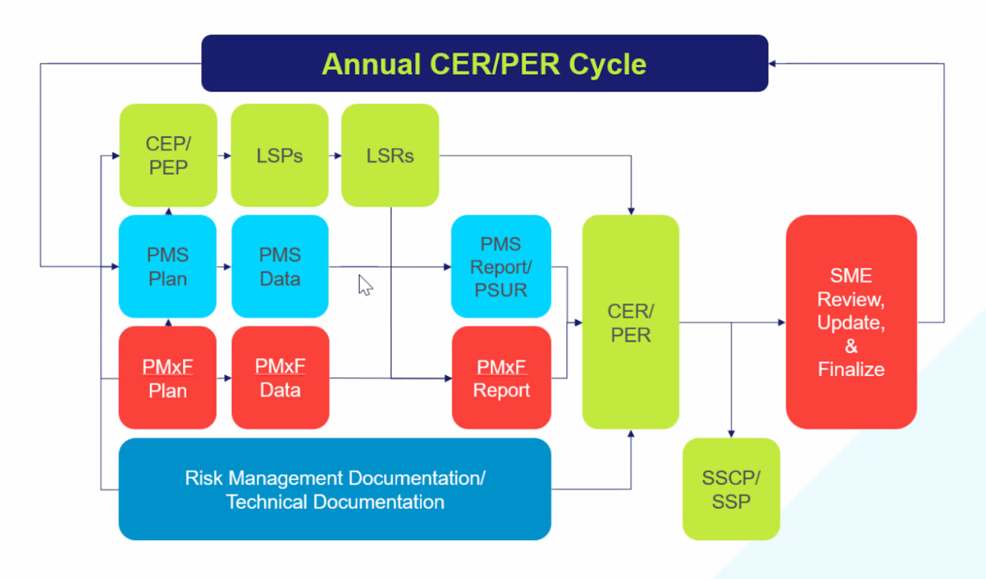
Understand the annual CER lifecycle and the roles of all stakeholders involved.
Clinical, quality, risk management, and any other team that participates in the lifecycle should create a cohesive strategy to ensure they are working in tandem to create consistent documentation. The silo approach will not work for MDR, and many manufacturers are learning this the hard way.
Create documentation concurrently.
Although you may not be able to create a complete document set at the outset, if you initiate individual documents at the same time and communicate along the way, your technical documentation is more likely to be consistent. The same is true for updates and any new information that is added after CE marking has been completed. When you have a cohesive strategy that includes a process for cross-referencing related documents, you can save hours of time correcting inconsistencies.
Have a data collection strategy.
Make it clear which groups are collecting the various types of data required, and have a strategy for data normalization, so teams are using the same terms for:
- Intended purpose
- Indications for use
- Device description
- Residual risks
- Similar devices
- Any other data points found in multiple systems
Understand the proper sequence so that all these data inputs feed into each other appropriately. It’s also important to make sure all the right people have access to the data they need to do their work, including database login credentials, knowledge of when access to data expires, and so on. Create a schedule so that data is periodically gathered and updated in a timely manner. A program director who works across teams to ensure the process is being followed will keep teams on track and maintain data integrity.
Common Mistakes Manufacturers are Making
With the transition to EU MDR, there are bound to be some stumbling blocks. By learning from the experience of others, you can avoid some of the most common mistakes.
They aren’t thinking ahead.
MDR compliance entails much more than just the initial application. It’s an ongoing process that requires a coordinated effort in order to do it efficiently and ensure there are no gaps. Having all parties aware of the overall process and involved from the beginning can prevent future issues. Manufacturers need to be thinking about what’s due in May 2022, May 2023, and beyond so they can manage resources for the work required.
They don’t involve Subject matter experts.
Subject matter experts should be involved throughout the entire process. Risk management, PMCF, regulatory, clinical, and post-market surveillance SMEs can and should contribute at key points before work is completed. By including SME review throughout the process, you can avoid costly and time-consuming corrections and notified body findings.
They don’t involve decision makers or have a clear escalation path.
The process of compiling technical documentation can uncover issues that need to be addressed at a higher level. For example, decisions to change the instructions for use or to start a new clinical investigation cannot be made by a medical writer. Ensure that key decision makers are informed along the way and have an opportunity to weigh in as needed.
Benefit from Our Unrivaled Collective Knowledge
Our “Best Practices and Processes for Integrating CERs and Post-Market Surveillance Under EU MDR” webinar reflects on common MDR certification scenarios to demonstrate how the best practices can be implemented in the real world. Because maintaining compliance is an ongoing process, we include recommendations for both initial application and maintaining documentation through the lifecycle of a device. A holistic program timeline, which clearly highlights the need for integration among teams, shows the cadence of technical documentation, who is involved, and when.
If you missed our webinar, “Best Practices and Processes for Integrating CERs and Post-Market Surveillance Under EU MDR,” it is now available on demand. Click here to get access now.


.svg "white RQM logo")




