
The first three Performance Evaluation Consultation Procedure (PECP) views have been published for a professional SARS-CoV-2 immunoassay and a professional nucleic acid testing (NAT) assay as well as a near-patient testing NAT SARS-CoV-2 product. Considering the actual criticality of these devices, these expert panel views are a big milestone to speed up conformity assessment work conducted by notified bodies (NB) to achieve In Vitro Diagnostic Regulation (IVDR) CE marking.
We will discuss specific challenges in the conformity assessment route for Class D devices and discuss the criticality – especially for SARS-CoV-2 devices.
Why is expert panel review needed for these devices?
CS under the IVDR are obligatory for the manufacturer to follow. But under the IVDR CS have not been published yet. However, the MDCG group has released a specific guidance, MDCG 2021-21 “Guidance on performance evaluation of SARS-CoV-2 in vitro diagnostic medical devices,” which should be considered as state-of-the-art requirements the manufacturer needs to apply.
The IVDR requires expert panel involvement for all Class D devices where no common specifications (CS) are available and the Class D device is considered to be the first certification of that type (Article 48 (6)). For this SARS-CoV-2 device, both requirements to involve an expert panel are met. There are no CS published yet and the product is considered to be the first certification of that type, which will be discussed here.
Expert panels are new stakeholders and strengthen the scrutiny mechanism for class D devices as required in Article 50 (see Appendix 1). [Expert panels are also required for high-risk medical devices such as Class III implantable and Class IIb active devices intended to administer and/or remove a medicinal product (combined devices). It should be noted that the timing for involving these expert panels differs significantly between IVDR and MDR. Under the MDR, expert panels receive the manufacturer documentation after the NB has had the chance to assess it. Whereas for IVDR manufacturers, that process occurs a lot faster and prior to NB review, as we will discuss later in this blog.]
The SARS-CoV-2 immunoassay device required expert panel involvement because there are no common specifications published yet and it was the first certification of that type of device.
Under IVDR, the NB is required to submit the performance evaluation report (PER) within five days of receiving it from the manufacturer. This implies that the NB itself did not do a conformity assessment on the PER and the overall technical documentation. The expert panel has 60 days upon PER receipt to publish their scientific view, which needs be duly considered by the NBs conformity assessment work, e.g., the review of clinical evidence and performance evaluation, according to Annex XIII.
As IVDR requires the expert panel view to be based on the PER only, it is vital that the manufacturer provides a comprehensive and thorough PER to allow third parties, such as the expert panel, to conduct an assessment that could lead to a positive recommendation.
Although the IVDR requires that only the PER is submitted to the expert panel, it is key for the PER to stand on its own as this is the only document that is reviewed by the expert panel.
For devices that require scrutiny by expert panels, it is therefore crucial that the PER contains more than an overall summary. It should include all key elements that allow traceability from performance claims to intended use/purpose claims, and be supported by objective and complete data, including a thorough evaluation of clinical evidence.
For devices that require PECP, RQM+ recommends supplementing the PER with the following documentation sets:
- Instructions for use (IFU)
- In-depth discussion of the state of the art
- Comprehensive analytical performance report, including information on the study design, methods and the relevant output data as applicable meeting Annex XIII 1.2 and 1.2.2
- Comprehensive scientific validity (SV) report, including a justification for the sources selected as per Annex XIII 1.2.1. It should also include a detailed review of those SV sources, including both favorable and non-favorable data as applicable.
- Comprehensive clinical performance report that includes details on the study design, methods and all relevant study data as applicable meeting Annex XIII 1.2 and 1.2.3
- Post-market surveillance (PMS) data:
- For “legacy devices” PMS data, e.g., experience from clinical use, should be covered and may be used to further support clinical performance as “other source of clinical data” and can be included in relevant sections of the PER. Ideally, PMS data will have been obtained from surveillance data in Europe. When relying solely on other jurisdictions, a solid justification will be required.
- For new devices, (e.g. SARS-CoV-2 tests), “PMS” data (e.g. monitoring of current best practice/acknowledged state of the art) should be included to demonstrate established clinical evidence and clinical benefit at the time of design verification and validation activities. Does the SARS-CoV-2 NAT assay detect today’s dominant virus mutation? These PMS data can be included in relevant sections of the PER/state-of-the-art discussions.
What are CS?
CS are defined as a “set of technical and/or clinical requirements, other than a standard, that provides a means of complying with the legal obligations applicable to a device” (Article 2 (74)). CS cover specific requirements, such as sensitivity/specificity, to be achieved by the device and clear instructions for sample panel selection (number, types). The CS follow the same principles as common technical specifications (CTS), which are mandatory under the IVDD 98/79/EC.
Although CS are mandatory requirements for class D devices under the IVDR, they are yet to be published in the Official Journal of the European Union. In the absence of CS and specifically for SARS-CoV-2 the Medical Device Coordination Group (MDCG) has released MDCG 2021-21 “Guidance on performance evaluation of SARS-CoV-2 in vitro diagnostic medical devices” which should be considered as state-of-the-art requirements for the manufacturer to apply. It is expected that the requirements in MDCG 2021-21 will form the basis of the future SARS-CoV-2 CS.
Once published in the OJ, SARS-CoV-2 common specifications become legal requirements. Nevertheless, as new evidence on the virus emerges it is likely that those CS will naturally evolve over time. This may be done in future through further revisions of the current MDCG guidance rather than immediate updates to the CS as these have to go through a legal pathway for publication mandated by the EU Commission. As a result, all manufacturers of SARS-CoV-2 devices need to be aware of any new MDCG guidance on that subject, which might affect your claimed clinical evidence and clinical benefit. Does the SARS-CoV-2 assay have a positive impact on patient management and public health for today’s dominant virus mutation?
The NB will require proactive monitoring on PMS data, e.g., clinical data, in particular for SARS-CoV-2 products, as well as clear triggers and provisions for conducting a Post-Market Performance Follow-Up (PMPF). Don’t wait for additional MDCG Guidance or CS to be published. You need to demonstrate the state of the art for your specific device, which is challenging with this continually changing virus.
What is "first certification of that type"?
There have been some confusions around the term “first certification of that device.” MDCG has published the guidance MDCG 2021-22 “Clarification on ‘first certification for that type of device’” and corresponding procedures to be followed by notified bodies, in context of the consultation of the expert panel referred to in Article 48(6) of Regulation (EU) 2017/746, to provide more clarity on what this term means.
First certification of that type refers to all devices that differ in two main aspects – first in the intended purpose and second in the analysis technology – to already CE-marked devices under the IVDR and In Vitro Diagnostic Medical Devices Directive (IVDD). Under the IVDD, only a limited number of products have been CE-marked as high-risk devices. List A under Annex II (e.g., HIV, hepatitis B virus, hepatitis C virus, some blood grouping products will fall into Class D devices under the IVDR. As a result, multiple high-risk Class D devices will be the first certification of that type as these have not been in the scope of the IVDD (e.g., SARS-CoV-2 devices, syphilis assays for donor screening, as well as other products used for in donor screening to detect transmissible agents).
The three SARS-CoV-2 devices represent distinguished SARS-CoV-2 product types and each product has been recognized as first certification of that type.
The SARS-CoV-2 devices vary in their intended purposes and analysis technology. For example, in the intended user (professional versus near-patient testing), in the function of the device (detection, differentiation versus screening), in the target genes for the both NAT devices, in the used specimen types (serum, venous plasma versus nasopharyngeal swab) and in the analysis technology, immunoassay versus NAT.
Having the first expert panel view for a device that is the “first certification of that type,” the manufacturer needs to assess if their SARS-CoV-2 devices are still considered to be the first certification of that type. Being considered as a “similar” device, the manufacturer needs to consider the claimed intended purpose according to the elements listed in the MDCG guidance – MDCG 2021-22, which are traceable to elements of GSPR 20.4.1 (c). In addition, the type of assay technology should be verified to determine if it is similar to your product.
So far, the first three PECP views on a SARS-CoV-2 devices have been issued but no IVDR CE certificate has been issued yet. But how can NBs avoid multiple PER expert panel consultations for similar SARS-CoV-2 devices?
MDCG 2021-22 requires NBs to identify similar devices up front to avoid submitting these PERs to the expert panel. They want NBs to wait until the expert view is published for the first certification of that type, then incorporate this view in their conformity assessment work for similar class D SARS-CoV-2 products.
Although we do not yet have a fully operational regulatory infrastructure to ensure a continuous supply of in-vitro-diagnostic devices to all healthcare systems, it is promising to see that the pace of the Class D conformity assessment progress is increasing and protecting patients by establishing greater scrutiny for these critical high-risk devices.
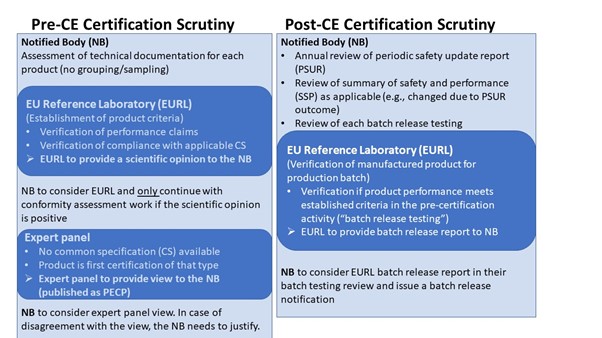
Appendix 1 – Note on Scrutiny for Class D Devices
The IVDR scrutiny mechanism for Class D devices applies both on pre CE-certification activities before a CE certificate is issued and post CE-certification activities to maintain the achieved CE certification. Pre CE-certification activities include consultation of additional stakeholders, EURL (EU Reference Laboratories) and the expert panel as applicable. Post CE-certification activities include continuous involvement of EURL for batch release testing as applicable. [Note: not all elements of the Class D scrutiny process, e.g., the involvement of EURL, have been discussed in detail here. More to come!]
How can RQM+ help you?
The conformity assessment for high-risk Class D devices is particularly challenging as additional stakeholders are included. We can support you in your regulatory strategy and provide clarity on the additional stakeholders needed for these critical, high-risk devices, considering the new progressive rollout for Class D devices to be compliant by May 2025.
RQM+ supports you from the start to achieve timely IVDR CE-marking, helping with communication and pre-application activities, as well as setting up an IVDR-compliant QMS and technical documentation for your entire product portfolio.


.svg "white RQM logo")




