




What is regulatory intelligence?
Ok, so what on earth are we talking about when we say regulatory intelligence? There are no formal definitions for regulatory intelligence in regulations or standards relating to medical devices or in vitro diagnostic devices. Without delving into the multitude of uses and dictionary definitions for ‘intelligence’, we can summarise them with some key phrases:
- Information gathering
- Understanding information
- Applying the learnings
Regulatory intelligence is often talked about within the pharmaceutical industry and its usage frequency in the MedTech industry is steadily increasing. By amalgamating various definitions, we can understand regulatory intelligence (for the MedTech industry) to be:
“…a systematic process of collecting, analysing, interpreting and disseminating information about regulatory requirements, policies, and guidelines that affect the development, manufacturing, distribution, and surveillance and regulation of medical devices and IVDs.”
And “what is it for?”, I hear you ask. Well, the information gathered and insights gained can be used to inform decisions on business strategies (e.g. new products, new markets, new claims, favourable locations for study sites), to improve the overall effectiveness of regulatory submissions (e.g. avoiding common mistakes, reacting to new guidance, more accurate planning and budgeting), or to mitigate risks to regulatory compliance and/or business continuity, or to identify new or growing areas of technical expertise (e.g. where your organisation needs to bolster or improve competence in a technical subject such as cybersecurity or machine learning).
Context is Critical
The key steps in the regulatory intelligence process are illustrated in Figure 1.

The act of gathering information includes collecting and analysing regulatory information from various sources, including regulatory agencies, industry associations, manufacturers, competitors, academics and scientific literature. That could be an endless task, so the context of your activities is critical to the effectiveness of your process, e.g. the nature of your business, your products and/or services, your competitors, the markets in which you operate, your customers, your future plans. If you fail to clearly identify the scope of your process, and tailor your methods, etc. accordingly, then you will find that you struggle to see the wood for the trees (or the forest for the trees, if you prefer) and your process seems inefficient and ineffective (Figure 2).

To make the information collection relevant for your organisation and objectives, you should understand and define the scope of your process:
- The types of information to be collected,
- The subjects/topics relevant to you,
- The sources of that information,
- The availability of that information,
- The frequency that the information is made available.
The application of these constraints and filters can be visualised similar to the illustrations of the swiss cheese model, often used to show the effect of combining multiple layers of imperfect control measures (Figure 3). By restricting and filtering the information gathered, it makes the analysis and triaging of the information much more manageable. The information can then be assessed for relevance, significance and urgency.

An ounce of action is worth a ton of theory
You now know what is changing or what is incoming or what has happened. Do not just sit on the information gathered; if it is relevant, what is it to be done about it?
Again, the context of your organisation is crucial. Does it affect regulatory compliance? Is it imminent? Which products/services are affected? Which markets are impacted? What is the scale of impact on products/services? What kind of work will you need to do in response?
Then it simply becomes an action management activity:
- define your actions clearly;
- ensure that they are measurable and achievable;
- assign an action owner and a feasible completion date;
- ensure that the required resources are identified and made available.
Depending on the size of organisation and the preferred decision-making processes, the need for action may be agreed quickly and then disseminated to the applicable personnel, or a proposal for action may be put to a panel of decision-makers to decide whether they agree with the proposal (and the assessment) before communicating the agreed actions to the applicable personnel.
If products, regulated activities or quality management system processes are impacted then the action management could fall within your organisation’s corrective and preventive action process. This depends on whether a nonconformity has already occurred due to the event or could occur if action is not taken before the event. Some examples are provided below:
Example 1, publication of ISO 14971:2019, provides a non-exhaustive illustration of a high-impact event that would need to be managed within the scope of the quality management system. In this example, the thorough analysis done at the outset, lays the groundwork for a detailed and prioritised quality plan. This attention to detail and planning goes a long way in demonstrating control within your quality management system.
No regulatory authority or conformity assessment body will expect major updates like this to be implemented immediately across all affected products, but they do want to see that you understand what is needed, have a plan with resources in place to execute the changes, and have prioritised the workload proportionate to risk (Note: they may disagree with you on this front!). Leaving these updates until the last minute or until they are requested / demanded, is not prudent as it typically results in audit findings or delayed submissions, for which the timeline for completion moves away from your immediate control.
Example 2, announcement of a proposed amendment to the US Quality System Regulations, shows a non-urgent low-impact event, that may turn into a high-impact situation later. This example highlights the benefits of active monitoring of the regulatory landscape even for non-urgent topics. Being aware of the proposal, the narrative around the proposal, the feedback from other stakeholders, etc. provides a good foundation to make preliminary judgements on what work will be required for implementation further down the line when the proposal becomes regulation. It enables informed decisions to be made when planning for resources and the timings of other activities. Being unaware of the proposal could leave you with a sudden, unplanned resource demand that negatively impacts the success on other business projects (e.g. submissions, site certifications).
| COLLECT | ISO 14971:2019 was published in December 2019 by ISO, quickly followed by EN ISO 14971:2019 and the national adoptions of those standards. |
| TRIAGE | Consider: What is the impact on the QMS? What is the impact on products already on the market? What is the impact on products currently in development and/or in the regulatory conformity assessment process? How critical is conformity to ISO 14971:2019 to regulatory compliance, competitive products and meeting customer expectations? Have the applicable regulators made any announcements about the new version? (e.g. has the EN version been harmonised for the EU? Have the FDA recognised this consensus standard?) Have the applicable regulators given any requirements or expectations for implementation? |
| ANALYSE | Review the 2019 version. Compare with ISO 14971:2009 and EN ISO 14971:2012. Identify any: new requirements; removed requirements; modified requirements, new/removed/modified informative guidance. What are the (high-level) differences between the requirements of the 2019 version and your risk management process and procedures? In general, what kind of updates will be required to risk management Files to bring them up to the level so that conformity with ISO 14971:2019 could be maintained. Do all existing risk management files conform to your current risk management procedure, or do they differ depending on age and product origins? |
| DISSEMINATE | Share with internal stakeholders; include them in the action-planning. |
| ACT | Document a gap assessment of your risk management process and procedures against ISO 14971:2019, including examples from existing and current risk management files. Plan for and document the changes needed for the risk management process and procedures, and how those changes will be communicated to those responsible for creating and/or maintaining risk management files and all other personnel involved in the risk management process. Update your risk management process and procedures (this would typically be seen as a corrective action, i.e. procedure is no longer conforming to the standard to which it claims conformance). Develop and document a plan/protocol for how to upgrade/remediate the existing risk management files (i.e. a plan for how implement the updated procedure and correct the risk management files…a correction). Include the methodology for what to do if new risks or ineffective controls are identified. Include criteria for escalating issues to management. Include prioritisation of risk management files based on:product classification, regulatory compliance risk (e.g. time to next submission or audit), commercial importance of product, lifecycle management status (e.g. if design change is planned this would be more urgent to update than a product that is intended to remain static for a few years), state of the existing risk management file (compared with current process). Roll out training to identified personnel to ensure that the updated procedure and the correction plan can be implemented effectively. Implement the correction plan/protocol and update the individual risk management files accordingly. |
Example 2: Amendments to the US Quality System Regulation (QSR)
| COLLECT | FDA publish final rule for amendments to the Quality System Regulation |
| TRIAGE | Consider the impact on existing products on the US market, planned submissions for the US, the sites responsible for those devices, scheduled audits, and likely FDA inspection dates. There is a two year time frame for implementation. |
| ANALYSE | What are the amendments? What are the differences between the old and the new? How do the amendments relate to the current QMS? What are the high-level differences between the current QMS and the requirements of this proposal? Which sites / product portfolios are affected by the amendment? How will the amendment impact on inspections methods, etc.? Are some sites or quality systems more at risk of non-compliance than others? E.g. they are not currently certified for ISO 13485:2016. |
| DISSEMINATE | Notify all internal stakeholders, especially for those responsible for quality compliance, and include the topic in upcoming Management Reviews. |
| ACT | Start a gap assessment on the amended rule. Establish a quality plan on the back of the results from the gap assessment; identifying actions, priorities, action owners, due dates, required deliverables. Continue to monitor FDA communications for updates and further news. Monitor other sources of insight (e.g. blogs, webinars etc.) from industry experts for their insights into interpretations. |
Sounds a lot like Post Market Surveillance to me!
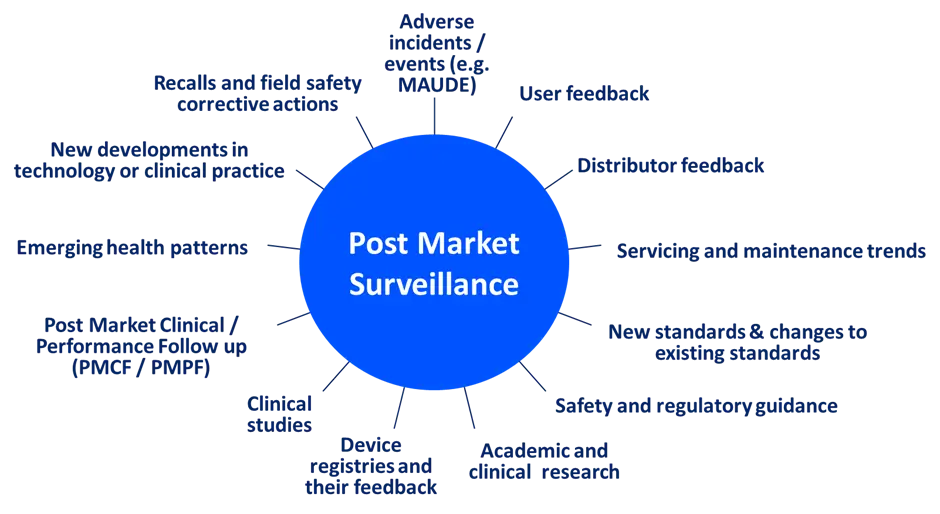
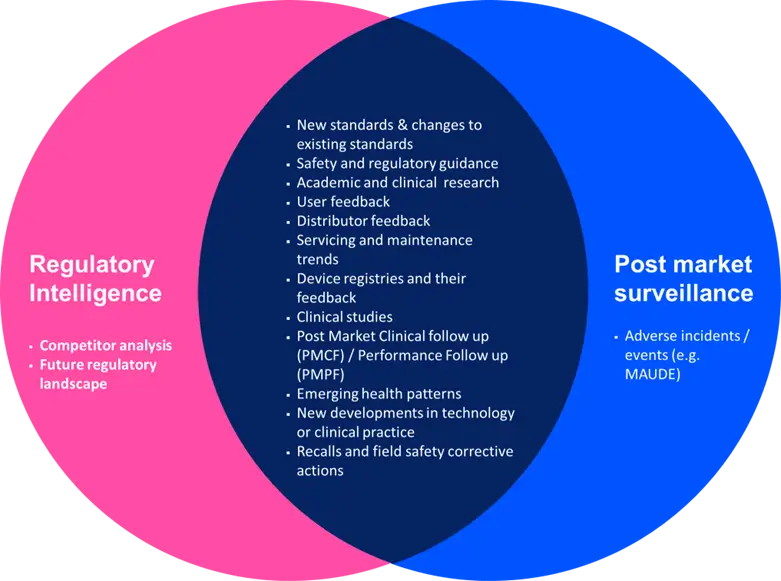
Post market surveillance should look for data from both internal and external sources. Figure 4 highlights some of the areas to be considered for external data (Note: so does not include sales data, internal non-conformance or corrective action data). For a device manufacturer, there is probably a significant amount of overlap between the information gathered and analysed for regulatory intelligence purposes and for product-specific post market surveillance activities (Figure 5). The scope of the two processes may be the same or similar (because they are both based on your organisation’s context) and the process steps and tasks may be almost identical, but the objectives of the two processes differ.
Post market surveillance is about monitoring the safety and performance of your device via proactive and reactive data collection methods, followed by analysis and evaluation of the collected data (or the ISO 13485:2016 definition if you prefer: “systematic process to collect and analyse experience gained from medical devices that have been placed on the market”). As described earlier in this piece, regulatory intelligence is unlikely to be product-focused (unless you only have one product in your portfolio), but it is intended to help inform business strategies and decisions. These decisions may be product-related, such as changes of direction for launch strategies or new ideas for lifecycle management of a product. Alternatively, it may relate to commercial activities, or operational decisions that may span all or multiple products, or may be independent of products completely.
Does that mean that having a post market surveillance process negates the need for a regulatory intelligence process? No.
As said above and shown in Figure 5, these two related processes have a lot of similarities but their respective objectives are different. That means that the same information can be reviewed but with two different mindsets. For post market surveillance the focus is simply on the product or product family: what does this mean for this product? For regulatory intelligence the focus is broader and is looking at all products and commercial activities; it is looking at the horizon to avoid compliance issues in the future; it is scanning data to seek out commercially advantageous regulatory strategies.
But there is more to regulatory intelligence than just an alternative take on post market surveillance.
Proactive regulatory intelligence as part of product development
Up to now, we have focused on the reactive and proactive collection of information on a routine basis, but regulatory intelligence can also be used for a planned collection activity with a very specific focus. A good example of this is the research done to build a regulatory strategy that informs the design and development plan and the commercial launch strategy (see Example 3 below). Key elements to consider include: applicable regulations, product classifications, applicable reimbursement codes, suitable claims to target, claims to avoid, known safety issues and failure modes, applicable testing schemes, and applicable safety standards to apply. This research is also very similar to that performed to understand the generally acknowledged state of the art as part of a clinical evaluation or performance evaluation (as required per applicable regulations).
Example 3: Building a regulatory strategy
| COLLECT | Collect data on similar devices, competitor devices and predicate devices already on the market, including from: Registration databases such as the FDA’s and EUDAMED, Clearance/approval documentation from the FDA, Incident databases such as FDA’s MAUDE, IFU, marketing literature and manufacturer’s websites. Freedom Of Information (FOI) requests from the FDA, Other publicly available documents such as the Summary of Safety & Clinical Performance (SSCP) or Summary of Safety & Performance (SSP) for higher risk devices on the EU market. |
| TRIAGE | What information is applicable to the existing product plans? How does the information gathered change the existing plans or preconceptions? Is urgent course-correction required on the development project? |
| ANALYSE | Are there alternative routes to market? What are the criteria to be considered, or risk mitigations needed, in order to decide on which route is preferred? |
| DISSEMINATE | Inform the relevant project team and responsible leadership of the findings and recommendations. |
| ACT | Build your regulatory strategy incorporating all the available public knowledge. Make recommendations based on the acceptable risk and reward balance. Identify areas for further information collection and/or ongoing intelligence monitoring to refine and improve the regulatory strategy. |
Do we need a procedure for regulatory intelligence? Should it be part of our QMS?
In short, yes and yes (for the routine intelligence gathering at least).
Many medical device manufacturers have a quality management system conforming to, or at least based on, ISO 13485:2016. The objectives, purpose and methods for regulatory intelligence should address elements of maintaining quality management system effectiveness, quality management system planning, management review, product requirements and the general monitoring and measurement activities. Having a documented procedure as part of the quality management system provides assurance that specific tasks and deliverables have been identified, that roles and responsibilities have been assigned and that interfaces with other quality management system processes are understood (e.g. with the CAPA and change management processes).
If there is no formal process or it is considered a business process rather than a quality system process, then there is a good chance that it will not be implemented or monitored as it would when under the focus of the quality management system. If that were to happen, key changes or events may be missed or seen too late, which negatively affects planning for quality system changes or product submission strategies. If the action requires identification and resourcing of specific competence from outside the organisation, ineffective intelligence gathering could mean that you are at the back of the queue when it comes to choosing talent.
What does the regulatory intelligence process look like within RQM+?
We don’t design, make or sell medical devices, our clients do. Our product is the services we provide to our clients. That is the lens through which we implement our regulatory intelligence process. Given that we have many different clients with varied product portfolios, that means our net is cast far and wide to try and ensure we don’t miss anything useful or beneficial. Whilst we have specific personnel who are responsible for the bulk of the intelligence gathering and analysis, it is also a collective effort with all personnel able to share information with the wider teams.
We analyse the information with the following questions in mind: What is the potential impact for:
- Our clients?
- Our prospective clients?
- Our current projects?
- Our planned projects?
- Our processes, templates, and tools?
- Our future business strategies?
- Our marketing strategies?
Once analysed and triaged, we share the information critical to our teams in proportionate time with an appropriate level of detail. This also usually includes archiving information in our internal Collective Knowledge Centre, a central repository for all things useful when it comes to MedTech, at least for regulatory affairs, clinical affairs and quality assurance. The aim here is to ensure that our teams have the relevant intelligence information and insights available at their fingertips, ensuring that they are not surprised if an existing client suddenly asks, “Have you seen that proposed amendment to the regulation? What do you think it means for us?”. It also enables our consultants to find guidance documents or standards by subjects or product areas.
It is also appropriate to be transparent here; clearly, we use these intelligence insights to position ourselves advantageously for new clients, new projects, etc. as do all other successful service providers. Also, some of these insights are made publicly available for everyone via our RQM+ Live shows, our technical blogs, our other website content and our LinkedIn posts.
More RQM+ resources at your fingertips:
For those eager to deepen their understanding and enhance their strategic approach, RQM+ offers a wealth of educational resources designed to elevate your regulatory intelligence capabilities. From detailed guides to insightful webinars, our resources are tailored to empower your team with the knowledge and tools necessary to leverage regulatory intelligence effectively.
Explore our offerings and join the community of MedTech professionals who are already benefiting from the competitive advantage that RQM+ educational resources provide. Embrace the power of regulatory intelligence with RQM+ and drive your organization towards greater innovation, compliance, and market leadership.
- Watch RQM+ Live! #76: MDCG 2023-7: New Clinical Evidence Pathways for Legacy and New Devices
- Watch RQM+ Live #75: Exiting PFAS: A Strategic Blueprint for Medical Device Manufacturers
- Follow #MedTechVoices on LinkedIn and get insights from our team as they analyze each of the opinions published by the Expert Panels as part of the Clinical Evaluation Consultation Process.
- Follow RQM+ on LinkedIn for all of our updates!
- Nancy Morrison’s Keys to Thriving in Regulatory Affairs: Lessons Learned from a 30+ Year Career