



Introduction
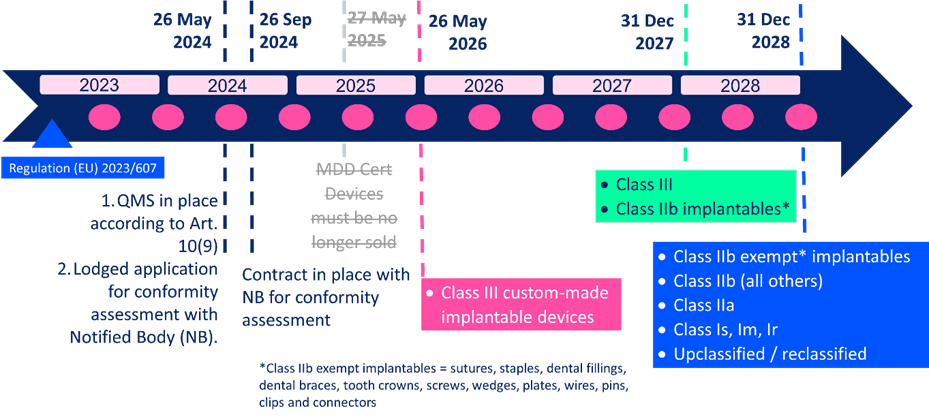
Regulation 2023/607 removed the May 2024 transition deadline for medical devices transitioning from MDD/AIMDD to MDR 2017/745. The end of the transition period was pushed back to 2026, 2027 or 2028, depending on the classification of the device.
The collective sigh of relief amongst many device manufacturers could be heard from Athens to Zaragosa! An extension of 3 ½ or 4 ½ years for the majority of transitioning legacy devices sounds great, but that time needs to be used wisely, and there are a couple of earlier deadlines that need to be met in order to make use of the extensions.
We’ve picked out the key dates and considerations for legacy device manufacturers in this blog post. For even more insights and directly from notified body BSI, sign up for our RQM+ Live! show on 27 July 2023, where BSI’s Head of Clinical Compliance, Richard Holborow, will join us to provide a notified body’s view on the latest best practices for MDR compliance. Here’s the link to register.
1. Step back and reassess
The amendments to the transitional period brought about by Regulation 2023/607 may have opened some doors that were previously thought shut. CE Certificates that were valid on 26 May 2021 but expired before 20 March 2023 have a new lease of life if there was a signed agreement with a Notified Body for a MDR conformity assessment. The introduction of the ‘substitute device’ concept may also reinvigorate plans for legacy devices that are due to be replaced by new design iterations.
Remember…
Plans are living documents that should be revisited and updated in light of new information.
2. Know your dates
Whilst you may keep your Class IIa device on the EU market up until 31 Dec 2028 under these new transitional arrangements, it does not come for free. There are two key dates that form part of the conditions for the extended transitional arrangements. If the requirements have not been fulfilled by these two dates, MDD/AIMDD CE certificates (see also self-declarations for those devices up-classified under MDR 2017/745) will not be valid.

Ensure that you:
- Assess your QMS against Annex ZA of EN ISO 13485:2016 / A11:2021.
- Incorporate the QMS requirements of MDR 2017/745 into your internal audit schedules and plans.
- Assess your procedures and infrastructure so that you can readily support hybrid audits to give you more flexibility with scheduling. Both the MDCG and Team NB have produced position papers on the use of hybrid audits.
- Liaise with your, existing or new, notified body to understand what you need to provide as part of their application process.
3. Certificates please!
Knowing what the amendment to the regulation says is one thing, but proving your legacy device can legally remain on the market when your CE certificate appears to have expired is another. There will be no new certificates with new expiry dates issued by notified bodies under the MDD/AIMDD, as the extension of a certificate’s validity is done automatically by law as long as the conditions for extension are fulfilled. The simplest way to demonstrate the extended validity (e.g. to customers) is via a self-declaration; confirming that you fulfil the conditions and requirements of MDR 2017/745 Article 120(3) (as it was amended by Regulation 2023/607). Fortunately, MedTech Europe, COCIR and others have produced a template for legacy device manufacturers to use: Manufacturer’s declaration in relation to Regulation (EU) 2023/607. This self-declaration also acts like a checklist of what you, as a manufacturer of legacy devices, need to do in order to make use of the extended transitional period.
Additionally, you can support your self-declaration with a confirmation letter from your notified body (i.e. the notified body who will be conducting your conformity assessment under MDR 2017/745). This confirmation letter covers the scope of your certificate(s) and the content needs to be verified by your notified body. Be advised: this confirmation letter takes time to prepare and is unlikely to be produced within days of your request.
Ensure that you:
- Identify and include all of the relevant devices and codes in the scope of your self-declaration (and notified body confirmation letter). Accidental omissions from these documents may prevent efficient sales to new customers or registrations in other markets.
4. Get your documents in order
Once your contract is in place with your notified body, you can schedule the assessment of your technical documentation. One new addition to the process is the notified body completeness check, performed by ~81% of the Team NB members (Team NB, Medical Device Survey 2022). The 2022 survey data presented by Team NB indicates that the submissions were significantly lacking in the required content (i.e. as per Annexes II and III of MDR 2017/745), i.e. 80% of those notified bodies performing the checks reported that technical documentation was only 50% complete or less. This tallies with the opening remarks in the Team NB best practice for MDR technical documentation. In that guidance, Team NB state the most common reasons for delays in the reviews are due to incomplete submissions (required evidence not provided) and/or the lack of a cohesive structure (evidence is provided, but is not easy for the reviewer to locate). Anecdotally, delays in reviews are being seen where manufacturers are unprepared for answering the reviewers questions, and so new evidence needs to be prepared rather than supplying existing objective evidence.
Consider:
- Familiarizing yourself with the Team NB guidance and the applicable MDCG guidance. While these are not mandatory, they provide the insight into what the notified body reviewer(s) will be looking for in your evidence.
- Assessing your technical documentation thoroughly before submitting to your notified body; preferably use an independent reviewer, i.e. outside of the specific project team (either internal or external).
- The questions you may be asked by your reviewer and be ready to provide that information (e.g. up to date post market surveillance data, evidence of active risk management activities).
5. Time is on whose side?
The 3 ½ or 4 ½ year extensions for the validity of legacy device CE certificates are intended to help notified bodies get through the required conformity assessments. It was not primarily intended to give device manufacturers more time to compile their evidence for compliance to MDR 2017/745.
The contract with your notified body needs to be in place by 26 September 2024. By that point, you need to be clear with your notified body when your technical documentation and QMS will be ready and available for the conformity assessment.
The data presented by Team NB indicates that notified body reviews of technical documentation under MDR 2017/745 are mostly taking up to 18 months. That does not necessarily mean that the review can safely take place in 2026 or 2027 in order to be complete before the 2027 or 2028 deadlines, respectively. Throughout the transition period and up until the CE certificate is issued under MDR 2017/745, your legacy device is restricted. There can be no significant changes in the design or intended purpose of the device, otherwise, it invalidates the extended validity of the MDD/AIMDD CE certificates. Manufacturers who successfully transition to CE marking under MDR 2017/745 earlier are in the position to make changes (still under the oversight of the notified body’s conformity assessment) to respond to changing market needs, customer requirements or technological advances when compared to manufacturers making full use of the extended validity of the MDD/AIMDD CE certificates.
Ensure that:
- The criteria for significant changes, as per MDCG 2020-3 Rev.1, are embedded in your change management processes.
- Planned changes are evaluated alongside your regulatory strategy and the timeline for review provided by your notified body.
Want to know more?
Sign up for our RQM+ Live! show on 27 July 2023 where BSI’s Head of Clinical Compliance, Richard Holborow, will be joining us to provide a notified body’s view on best practices for MDR compliance, including tips for manufacturers of legacy devices transitioning to the MDR. If you’re reading this and the date of the show has passed, please know it’s available on demand at the link.
If this post has helped you realized or otherwise inspired you to seek support at your organization, please contact RQM+ for any needs related to QMS implementation, independent QMS audits, compilation and/or remediation of technical documentation, pre-submission assessments of technical documentation, responding to notified body review questions and/or planning of post-market activities to gather more evidence.