




By Ed Ball, CEng, MIPEM, MIMMM – Manager, Intelligence & Strategic Execution
Introduction
This blog on off-label use is the second part, following a blog where I discussed the ins and outs of intended use. I looked at the various definitions for intended use in widely applied regulations and standards, decomposed the intended use into its various structural components, described the critical role that the intended use has within all things medical device and discussed how the intended use will likely evolve and change over time. I warned that we should not think of the intended use as a single statement but rather a more complex specification for how we intend a specific medical device to be used.
In April I presented at the 2nd International Conference on Medical Device Safety Risk Management in Amsterdam. My talk was about intended use, reasonably foreseeable misuse and off-label use. Why did I choose this topic? Firstly, because understanding and characterising the intended use of a medical device is so critical that I don’t think we can talk about it enough. Secondly, because I have noticed an increase in the discussions around off-label use, especially in the context of real-world data and EU requirements for post-market clinical follow-up (PMCF).
So, What Is “Off-Label Use”?
To discuss off-label use, you first need to know what is ‘on-label use’ (that is not a phrase I want to stick). This is the intended use. Therefore if the intended use is the path, laid out in the device’s labelling and instructions for use, then off-label use is the deviation from that path.
Now you would think there would be an official definition for off-label use. Of course there is. Where?
- In at least one of our favourite standards? Nope.
- Obviously by the IMDRF? No, afraid not.
- In our most widely used medical device regulations? Third time lucky? No, again.
The only ‘official’ place I could find an actual definition of off-label use was in the US regulation 32 CFR 199.2 which provides the definitions for the guidelines and policies for the administration of the Civilian Health and Medical Program of the Uniformed Services (CHAMPUS) for the Army, the Navy, the Air Force, the Marine Corps, the Coast Guard, the Commissioned Corps of the U.S. Public Health Service (USPHS) and the Commissioned Corps of the National Oceanic and Atmospheric Administration (NOAA). Just what you were expecting right?
How Is It Defined?
This well-known US regulation defines off-label use (of a drug or device) as:
A use other than an intended use for which the prescription drug, biologic or device is legally marketed under the Federal Food, Drug, and Cosmetic Act or the Public Health Services Act. This includes any use that is not included in the approved labeling for an approved drug, licensed biologic, approved device or combination product; any use that is not included in the cleared statement of intended use for a device that has been determined by the Food and Drug Administration (FDA) to be substantially equivalent to a legally marketed predicate device and cleared for marketing; and any use of a device for which a manufacturer or distributor would be required to seek pre-market review by the FDA in order to legally include that use in the device’s labeling.
The UK’s MHRA talk about off-label use in the context of advice for healthcare institutions and clinicians, warning of the safety risks and liability that come with such use. The MHRA do not however provide an official definition. Australia’s TGA provide their own online guidance on off-label use and it includes a simple definition as follows:
‘Off-label use’ is using a product for a reason not listed as one of the indications for use in the Australian Register of Therapeutic Goods (ARTG).
Whilst this TGA definition is simple it is also misplaced in my opinion. For those paying attention to my last blog, indications is but one part of the intended use. Therefore off-label use can occur on other elements of the intended use other than the indications. Perhaps it is down to how TGA define intended use and indications? That is a rabbit hole for another day.
In conclusion, my simplistic definition is “a use for which the device is not legally marketed, based upon the intentions communicated by the manufacturer”.
Perceptions of Off-Label Use
Off-label use is often seen as a negative thing. It is not uncommon for ‘off-label use’ and ‘user error’ (deliberate choice of ‘user’ there before anyone calls me out on it) to be used as defensive responses to incoming complaints about device failures or poor performance.
Why do we see it in such a negative context? Just like the advice in the MHRA’s guidance, many hear off-label use and are immediately concerned with increased risks to patients and users, or about unproven effectiveness of the device, or even about increased product liability for the manufacturer (or for the healthcare professional, or their institution).
Those with a more open, positive mindset may hear off-label use and think about opportunities to meet unmet clinical needs, to provide improved care options for patients, to improve user experiences and opportunities for future device development.
It’s crucial to consider both sides of this issue to manage this complex topic effectively.
The Spark That Lit the Fire
The trigger that made me think about off-label use more closely was the Team-NB position paper from 2022 “Data generated from ‘Off-Label’ Use of a device under the EU Medical Device Regulation 2017/745”. What was it that caused the spark? The logical narrative? The tidy formatting? The pretty logo in the header? All good guesses, but no it was this statement that really got me thinking:
“Foreseeable misuse may be identified through usability studies or pre-market clinical investigation reports but it is often difficult for manufacturers to predict areas of future misuse…”
The first part of that sentence suggests that we can identify ‘foreseeable misuse’ through usability studies or pre-market clinical investigations. This is true, but they are not the only weapons in our armoury. The second part of that sentence states that it is hard to predict reasonably foreseeable misuse. My stance is that in many cases, off-label use can be readily foreseeable, even in the pre-market development stages. To achieve this I don’t believe that we need fancy AI-enabled predictive models; we should be able to utilise the tools and methods that we currently have available, as well as channelling our inner 3-year-old and keep asking questions.
Identifying Off-Label Use
This will not come as a shock, but I am obviously going to come at this from a risk management perspective. Following ISO 14971:2019, one of our key steps in the risk management process is the risk analysis. To start this analysis, we document both intended use and reasonably foreseeable misuse of the device. But that’s not off-label use I hear you say! Wait for it.
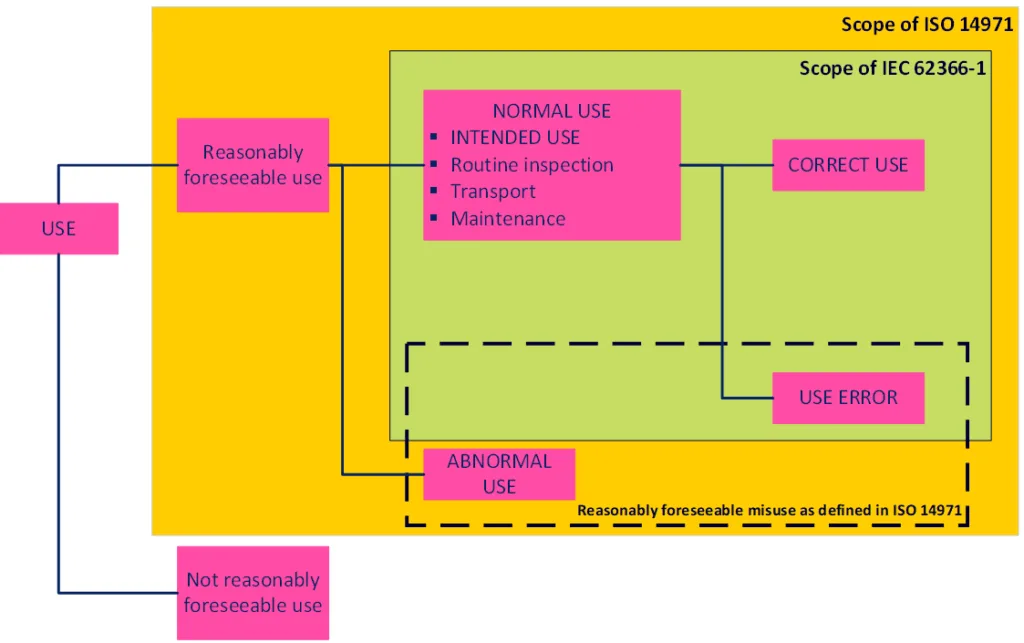
Figure 1 indicates the difference in scope of ISO 14971 and IEC 62366-1. Figure 2, also from IEC 62366-1, illustrates the links between the different types of use for a medical device. Neither mention off-label use, but both indicate that abnormal use is in scope of ISO 14971 and can thus either be part of the intended use or the reasonably foreseeable misuse. IEC 62366-1 provides some examples of abnormal use including the ‘conscious disregard for the contraindications’, which based on our previously identified definitions fits the bill for off-label use. MDCG 2023-3 Rev. 2 also states that abnormal use would include off-label use of a device.

By definition, reasonably foreseeable misuse can be foreseeable, meaning we can predict it based on human behaviour. It does not take a rocket scientist to look at the layout of the paths in Figure 3 to identify the potential for users of those paths to create the desire path that cuts the corner because the intended path directions are not optimal for the routine direction of travel.

We would typically start identifying reasonably foreseeable misuse before any usability studies or clinical investigations are commenced. Tools and methods for this prediction can include:
- Key Opinion Leader feedback: What do your expert users think?
- Customer feedback: Listening to what the customers really need, or what is missing from their treatment options.
- Brainstorming: Unstructured thinking through of the intended use and potential misuse scenarios.
- State of the art (SOTA) analysis: Analysing other treatment options for the intended use, analysing the intended use and contraindications of similar devices, consideration of the clinical context of the device and what other treatments and activities would be occurring in parallel or close proximity.
- PMS Data Analysis: Analyzing post-market surveillance data for instances of use error, reasonably foreseeable misuse and off-label use.
- Clinical Literature Analysis: Reviewing existing clinical studies and literature for reported cases of use error, reasonably foreseeable misuse, off-label use or indications of an unmet clinical need.
- Analytical techniques (e.g. task analysis): Logical step-by-step analysis of the discrete tasks involved with the use of the device and consider what could happen, e.g. considering the perception, cognition and action framing.
- Competitor analysis: Analysing the intended use and contraindications of their devices. If they have broader indications than your device, that is a sure-fire indicator that your device could also be used in those broader indications if they are seen by clinicians as equivalent devices.
- Usability Studies: Conducting formative and summative studies to identify potential errors.
- Exploratory clinical investigations: Conducting exploratory investigations may identify situations not anticipated by the designers and may highlight user needs or questions that were not previously anticipated.
After you’ve gone through at least one of these methods, what do those results look like? Table 1 below provides some examples of the kind of situations the above methods can help identify before the product is launched to market.
| Element of intended use | What we intend… | But what if it is used like this? |
|---|---|---|
| Intended medical indication(s) | For repair of weak soft tissue in the abdominal wall | Prophylactically used to strengthen tissue around surgical incisions |
| Intended patient population(s) | Adults (unspecified) | – Paediatrics, neonates – Pregnant women – >70 years old – Different ethnicities – Comorbidities |
| Intended part of the body or type of tissue applied to or interacted with | Intact skin | – Breached or broken skin – Nasal or oral mucosal surfaces |
| Intended duration of use | 14 days | – 25 days – 7 days |
| intended user profile(s) | Unsaid <fit and healthy adult> | – User with a prosthetic leg – Short / tall users |
| Healthcare professional | – Lay person | |
| use environment(s) | A State-of-the-art clinic | – Rural clinics – Domestic homes |
| Intended lifetime of the device | 10 years | – 14 years* |
| Intended accessories | Only used with our accessory | – Used with any accessory to hand |
| adjunctive devices &/or therapies | Immediate recovery period involves daily administration of medicine X | – Used with medicine Y – Lower dose of medicine X – Less frequent doses of medicine X |
By understanding these potential scenarios, manufacturers can better prepare for off-label use by making some important decisions.
Pre-Market Decisions
With each of these instances of reasonably foreseeable misuse identified, product development teams have a decision to make. Adopt this likely use into the intended use, or keep it very much in the misuse category.
If adopted, this aspect of the use needs to be added to the intended use (e.g. the use specification as per IEC 62366-1) and managed as a design change in the development project. Management of that change should include identification of any additional design input requirements to accommodate this aspect of use, any changes to existing design specifications (including information for use), any new hazards, hazardous situations, harms etc. to be considered in the risk analysis and any additional design verification and/or design validation work to generate the applicable objective evidence (e.g. laboratory testing, usability studies, clinical investigations, performance evaluation studies).
If the identified misuse is still considered misuse (i.e. not adopted) then it should be determined whether any further risk control measures need to be implemented (e.g. identification of contraindications) and then subsequently verified as effective.
As shown in Table 1, this is probably not a one-time thing. This will likely occur several times during the development of a medical device, and the intended use will be an iterative thing, with details added and excluded as decisions are made (Figure 4).

Ultimately, we have to settle on a defined intended use for the purpose of regulatory conformity assessments and market launch. But it does not stop there.
On the Lookout for Off-Label Use
Once a medical device is on the market, there are numerous requirements for the monitoring of the device’s performance and safety. This includes monitoring for off-label use. Those use situations categorised as misuse in the pre-market phase, should now be monitored to some extent to determine whether they are occurring in reality, and if so, whether they are resulting in the predicted outcomes. This means that the post-market surveillance system should be designed in a way to be able to detect instances of off-label use. One way to achieve this, is to use the IMDRF’s adverse event coding framework within the complaints handling system, which includes a specific code for off-label use as part of the codes for investigation conclusions (Annex D).

The description of off-label use in this IMDRF code (D1103) could be argued to be more practically useful than those definitions previously presented. However, in terms of the IMDRF’s codes, it is confusing because there are at least two other codes addressing specific instances of off-label use:
- Shelf Life/Expiration Date Exceeded
- Reuse of Single Use Device
Another route to identifying actual instances of off-label use is through post-market clinical follow-up (PMCF) or post-market performance follow-up (PMPF). It is important these follow-up activities ask the right questions to help identify off-label use. Asking “Did you use the device off-label?” may not be the best way to gather the desired information.
A third method could involve monitoring social media for information that indicates off-label use. This may not be logical or feasible for some devices, but should be considered for many devices that are long-term implants or require continuous use (e.g. a blood glucose monitor), where there may be various online groups for patients/users/carers to share stories and discuss ways to help manage their conditions etc. This may include untried suggestions of how a device could work better (e.g. “despite what it says in the instructions, I’ve found that it works better if I place the sensor on my upper thigh rather than my upper arm”). This kind of feedback can shed light on common complaints that are never reported directly to the manufacturer, as well as instances of off-label use and/or promotion of off-label use by individuals.
Another option is to look for off-label use during literature searches within post-market surveillance activities. This may require specific search criteria related to the nature of the off-label use (e.g. citing the specific contraindicated indication or patient population).
Post-Market Decisions
When off-label use is identified in post-market, we have another key decision point. But before that, there is an often-overlooked action. If there is clear off-label use, e.g. clear disregard for contraindications, or working outside of the defined intended use, then the device manufacturer should be clear to the user that they have used the device off-label. Without that communication, regardless of what decisions come afterwards, the manufacturer could be considered to be tolerating the off-label use and bypassing the applicable conformity assessment. It could also have implications for product liability should any patient and/or user subsequently be harmed as a result of such off-label use.
For clarity, if there are instances of actual use of the device in areas where the intended use has been poorly defined for the user, this may not be considered off-label use. Recalling the simplified definition from earlier, it is the use as intended by the manufacturer that is then communicated to the users. If the finer details of the intended use are not defined or communicated to the end-users, they cannot be accused of using the device off-label.
Instances of actual off-label use may:
- signify an unmet clinical need, e.g. an underserved clinical condition or a small patient population;
- highlight confusion between similar devices with differing indications and contraindications;
- indicate a predictable response to the removal of a similar device from the market, leaving a gap in the market for a specific intended use;
- represent a clinician’s decision on how best to treat their specific patient with the equipment options that they have available.
There should be a check of the risk management file to determine whether the off-label use was anticipated or not. If not, why not? Does it reflect a gap in the methodology used for risk analysis, or in the contextual awareness of the development team? If this type of off-label use was anticipated, were there any controls in place to address this use? If so, how effective were they? These are all sensible questions to ask yourself (before your competent authority asks them of you!).
Ok, back to decision time. We now have to decide what to do with the identified off-label use. Maybe it is a one-off, maybe it is a trend of systematic off-label use. Either way, we need to plot our next steps.
Option 1 is to embrace this off-label use and adopt it. This would require a change management activity to update the intended use and then implement that change accordingly, possibly requiring additional regulatory conformity assessment(s) before communicating the change to customers and users. In many cases, this would require the gathering or generation of additional objective evidence to support this extended intended use (e.g. a new patient sub-population, or a new indication). Ensure that this is managed correctly as design creep without appropriate regulatory ‘approval’ is a known issue, especially witnessed by the FDA in the US.
Option 2 would be to stick with the current boundaries of intended use and maintain the stance that this type of use is off-label use. This becomes more risk focused and depends on the risk associated with the off-label use. This means risk both in terms of the occurrence of such use and the potential consequences, either in harm or ineffective treatment. Are the existing risk control measures sufficient? Do these real-world events change the acceptability of the risk associated with the off-label use? Are further risk control measures necessary and if so, what form will they take and how will you measure their effectiveness? It may be that the intended use was too vague and the off-label use was not clearly prohibited in the instructions for use, or it was determined to be unlikely to occur; either way action (e.g. now clearly stated as a contraindication) may be warranted.
Conclusion
Off-label use is a complex but fascinating aspect of medical device safety. It is not something to be inherently scared of. By understanding the intended use, identifying foreseeable misuse, and analysing the data, manufacturers can make informed, evidence-based pre- and post-market decisions. It’s all about balancing opportunities with risks to ensure the best outcomes for patients and users. This is especially important when it relates to off-label use that falls into the ’orphan’ category (e.g. an orphan patient population or indication), and there can be an inclination to contraindicate, rather than adopt, as the quicker solution.
Related Next Steps
Go back and check out part 1 of this blog: Intended Use: Foundation stone, guiding light or just a box to tick?
Revisit our RQM+ Live show from 2023 “Beyond Indications: Managing Off-Label Use for Safety and Compliance”
- Need help with anything off-label use?
- Does your risk management process need assessing to ensure that it supports the identification of off-label use?
- Unsure whether you have sufficient clinical data to support specific indications?
- Is your post-market surveillance system capable of detecting off-label use?
- Need to optimise your PMCF strategy to ensure that you can gather clinical data on off-label use?
Contact us to discuss how we can support your efforts.
Bibliography:
For those interested in diving deeper, here are some key references:
- ISO 14971:2019 Medical devices — Application of risk management to medical devices
- ISO 13485:2016 Medical devices — Quality management systems. Requirements for regulatory purposes
- IEC 62366-1:2015/Amd 1:2020 Part 1: Application of usability engineering to medical devices
- EU MDR 2017/745
- FDA Guidance on Human Factors and Usability Engineering management systems. Requirements for regulatory purposes
- ISO/TR 24971:2020 Medical devices — Guidance on the application of ISO 14971
- ISO 20417:2021 Medical devices — Information to be supplied by the manufacturer
- ISO/TR 20416:2020 Medical devices — Post-market surveillance for manufacturers
- Applying Human Factors and Usability Engineering to Medical Devices (FDA, 3 Feb 2016)
- Guidance on applying human factors and usability engineering to medical devices including drug-device combination products in Great Britain (MHRA. V2, Jan 2021)
- AAMI TIR50: 2014/(R)2017 Technical Information Report Post-market surveillance of use error management
- Purpose and Content of Use-Related Risk Analyses for Drugs, Biological Products, and Combination Products Guidance for Industry and FDA Staff (FDA, Draft, July 2024)
- IMDRF/GRRP WG/N47 FINAL:2024 (Edition 2) Essential Principles of Safety and Performance of Medical Devices and IVD Medical Devices
- Data generated from ‘Off-Label’ Use of a device under the EU Medical Device Regulation 2017/745. (Team-NB, 5 Oct 2022)
- Off-label use of a medical device (MHRA, 1 July 2023)
- Understanding regulation of off-label use of medical devices (TGA, 23 Sep 2024)
- Communications From Firms to Health Care Providers Regarding Scientific Information on Unapproved Uses of Approved/Cleared Medical Products Questions and Answers (FDA, Jan 2025)
- Responding to Unsolicited Requests for Off-Label Information About Prescription Drugs and Medical Devices (FDA, draft, Dec 2011)
- Off-label Use of Medical Devices (Medical Device Network, May 2017)
- IMDRF/Registry WG/N46 FINAL:2018 Tools for Assessing the Usability of Registries in Support of Regulatory Decision-Making
- IMDRF/NCAR WG/N14FINAL:2017 (Edition 2) Medical Devices: Post-Market Surveillance: National Competent Authority Report Exchange Criteria and Report Form
- MDCG 2023-3 Rev. 2 Questions and Answers on vigilance terms and concepts as outlined in the Regulation (EU) 2017/745 and Regulation (EU) 2017/746 (Jan 2025)
- For further reading – 5 Key Elements of GMP in the Food Industry