




By Chris A. Parr – Principal, CRO
The EU is constantly publishing new legislation on a variety of topics that have the potential to impact medical devices and IVDs. Often these new laws have complex and lengthy transition periods spanning multiple years. In this blog we will review some of the new and evolving legislation that manufacturers need to be aware of to ensure compliance in the future.
EU Product Safety Legislation
To set the scene one must consider the underlying product safety legislation in the EU which has three components:
- General Product Safety Regulation (GPSR) – Regulation (EU) 2023/988
- EU harmonisation legislation (sectorial)
- Standards
The GPSR outlines the general framework for the safety of non-food consumer products in the EU. It acts as a safety net, covering products, aspects and risks not addressed by harmonisation legislation. It also ensures that all products on the EU market are safe for consumers.
Sector-specific EU harmonisation legislation outlines the characteristics and safety requirements necessary for certain products. Products placed on the EU market must comply with this legislation e.g. medical devices, IVDs, personal protective equipment and toys.
Standards are technical specifications adopted by a recognised standardisation body and compliance is not compulsory. There are four distinct types of standards1 as follows:
- International – adopted by an international standardisation body e.g., ISO
- European – adopted by a European standardisation organisation e.g., CEN or CENELEC
- Harmonised – adopted on the basis of a request made by the Commission for the application of Union2 harmonisation legislation
- National – adopted by a national standardisation body e.g. AFNOR
Horizontal Legislation?
Horizontal legislation is separate from product safety legislation and applies across multiple sectors or policy areas. It establishes general principles or rules that cut across various domains (e.g. competition law, environmental protection, consumer rights). The General Data Protection Regulation (GDPR) is horizontal because it applies to all sectors handling personal data. Horizontal legislation often provides the overarching principles (e.g. non-discrimination, consumer protection), while harmonisation legislation implements these principles in specific sectors. Harmonisation laws must comply with horizontal legislation. For instance, a harmonised product safety law must still respect horizontal consumer rights laws.
New and Emerging Legislation
Sometimes manufacturers overlook the non-medical device and IVD regulations in the naïve belief that they are not required for CE marking or that someone else in their organisation outside of regulatory affairs will take care of it later. However, a single declaration of conformity is required to be drawn up covering all applicable legislation at the time of CE marking. Therefore, a product can only be considered truly compliant when all applicable harmonisation legislation and horizontal legislation has been covered. Notified bodies for medical devices expect manufacturers to comply with relevant EU horizontal and harmonisation legislation, especially when it intersects with the MDR or IVDR.

Recent regulations with the potential to impact medical device and IVD manufacturers include:
- Batteries regulation (EU) 2023/1542
- AI regulation (AI Act) (EU) 2024/1689
- Packaging/packaging waste regulation (EU) 2025/40
- European Health Data Space regulation (EU) 2025/327
In the following sections we will cover each of these regulations in more detail.
Batteries Regulation (EU) 2023/1542
The batteries regulation is a new horizontal regulation that was published in the OJEU in July 2023. Some provisions of the regulation began to apply from the 18th February 2024 (the Date of Application) with the remainder being gradually phased in over a period of 5+ years. Conformity assessment, CE marking and Declaration of Conformity requirements applied from the 18th August 2024. The general date of repeal of the legacy batteries directive 2006/66/EC is the 18th August 2025.
The regulation establishes a comprehensive legal framework for the entire life cycle of batteries placed on the EU market. It applies to all types of batteries, including:
- Portable batteries
- Light means of transport (LMT) batteries
- Starting, lighting, and ignition (SLI) batteries
- Industrial batteries
- Electric vehicle (EV) batteries
All batteries must undergo a conformity assessment, and the requirements vary by battery type and capacity. Medical devices and IVDs typically (although not exclusively) may use or contain portable batteries. The requirements for producers of portable batteries include:
- Meeting collection targets for waste portable batteries
- Marking batteries with the symbol for separate collection
- Marking batteries with the appropriate chemical symbol if they contain > 0.002% Cd, or > 0.004% Pb
- Complying with extended producer responsibilities (e.g. registration & reporting)
- Complying with general battery labelling requirements
- Labelling rechargeable portable batteries with information on the capacity
- Labelling non-rechargeable batteries with information on the minimum average duration
- Marking batteries with a QR code
- Meeting minimum values for the electrochemical performance and durability
The regulation also introduces the concept of removability and replaceability and the concept of battery due diligence.
Batteries must be designed so they can be taken out of a device without causing damage to the battery or the product. This must be possible using commercially available tools. The requirement applies to portable batteries in products like electronics, medical devices, and construction tools. Once removed, the battery must be replaceable with a new one – either the original or a compatible model. Replacement batteries must be available as spare parts from the device manufacturer for at least 5 years after the product is last placed on the market.
Battery due diligence refers to a set of mandatory obligations for large companies to ensure that the raw materials used in batteries are sourced responsibly, with respect for human rights, labour standards, and the environment. Companies must adopt a due diligence policy aligned with international standards and have their due diligence policies and implementation systems independently verified by a notified body.
AI Regulation (AI Act) (EU) 2024/1689
The AI regulation (EU) 2024/1689 was published in the OJEU in July 2024. The Date of Application is the 2nd August 2026, and some provisions of the regulation covering unacceptable risk AI systems applied from the 2nd February 2025.
The EU AI Act lays down harmonised rules for AI systems that are placed on the market, put into service, or used in the EU. It also prohibits certain AI practices. It is a horizontal regulation and thus applies to AI systems regardless of whether they are covered by other EU legislation or not. Therefore, medical devices and IVDs that utilize artificial intelligence to any degree, may be in scope of the regulation. The regulation introduces four tiers of risk classification for AI systems, with proportionate control measures and monitoring activities. The four categories, examples for each and the type of control are shown in Figure 1.
Medical devices and IVDs incorporating an AI system are considered high-risk under the AI Act if they are classified above Class I under the MDR or Class A under the IVDR. For high-risk AI systems, conformity assessment, CE marking and Declaration of Conformity requirements will generally apply from the 2nd August 2027.
The requirements of the AI regulation apply to:
- Providers of AI systems, regardless of whether they are established/located within the Union or in a third country
- Deployers of AI systems that are established/located within the Union
- Importers and distributors of AI systems
- Authorized representatives of providers who are not located within the Union
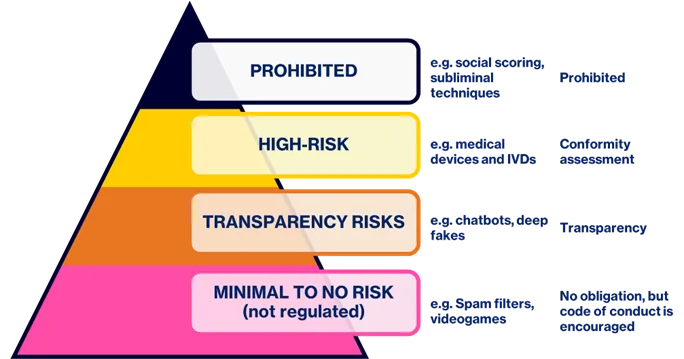
Medical devices and IVDs classified as AI systems under the AI regulation that are placed on the market before the 2nd August 2027, are exempt from the requirement to have a conformity assessment in accordance with the regulation. Any significant changes made to those devices after the 2nd August 2027, will trigger the need for a conformity assessment.
The regulation utilizes the Notified Body model which is not new to the medical device industry. It also introduces the European AI Board, comprised of representatives from the member states, which acts as a co-ordination platform and advisory body to the Commission (a bit like MDCG currently does under the MDR and IVDR). The regulation also creates a European AI Office which will be tasked with supervising the general-purpose AI models.
The conformity assessment required by the AI regulation can be combined with that for the MDR or IVDR and performed by the same Notified Body if they are designated under the AI regulation. The QMS requirements of the AI regulation can be combined into the QMS required by the MDR and IVDR. The recently released guidance from the Artificial Intelligence Board (AIB) and the Medical Device Coordination Group (MDCG) (AIB 2025-1 MDCG 2025-6) provides welcome guidance in this area.
Packaging/packaging waste regulation (EU) 2025/40
The packaging/packaging waste regulation (EU) 2025/40 was published in the OJEU in January 2025 (abbreviated as “PPWR”). The Date of Application is the 12th August 2026, and the legacy directive 94/62/EC will be repealed from that date. The new regulation is horizontal and applies to all packaging and packaging waste generated from all sources e.g. commercial, household, and industrial.
The PPWR is built on the extended producer responsibility scheme and is aligned with the EU’s New Circular Economy Action Plan (CEAP) which has the goal of making all packing reusable or recyclable in an economic way by 2030. The PPWR applies to all medical devices and IVDs. However, given the critical nature of medical devices and IVDs the obligations for manufacturers are significantly reduced compared with those for other sectors.
The intent of the PPWR is that packaging should be designed, manufactured and commercialised in such a way as to allow for its re-use as many times as possible or for high-quality recycling, and to minimise its impact on the environment during its entire life cycle and the life cycle of the products for which it was designed. The packaging of medical devices and IVDs is defined as “contact-sensitive packaging” under the PPWR. This exempts medical devices and IVDs from most of the requirements of the PPWR but not all of them. The main requirements affecting medical devices and IVDs are.
- Packaging minimization
- Labelling
- Re-use targets
- Extended producer responsibilities
Manufacturers must carry out a conformity assessment to ensure that packaging placed on the EU market complies with the regulation’s sustainability, recyclability, and labelling requirements. The regulation does not require CE marking on packaging.
European Health Data Space regulation (EU) 2025/327
The European Health Data Space (EHDS) regulation was published in the OJEU in March 2025. The Date of Application was the 25th March 2025.
The regulation established a common framework for the use and exchange of electronic health data across the EU and is considered harmonisation legislation. The EHDS is designed to support a single market for digital health services and products, fostering interoperability and innovation in electronic health record (EHR) systems. It enhances individuals’ access to and control over their personal electronic health data, while also enabling the secure reuse of this data for research, innovation, policy-making, and regulatory activities.
Primary and secondary use of electronic health data is established by the regulation. Primary use is to support healthcare delivery by enabling individuals and healthcare professionals to access, control, and share personal electronic health data across borders within the EU. Secondary use refers to the reuse of health data for purposes beyond individual patient care e.g. research, innovation, and policy making.
The EHDS requires all electronic health record (EHR) systems to comply with the specifications of the European electronic health record exchange format, ensuring that they are interoperable at EU level. The EHDS will also provide researchers and policymakers with access to specific kinds of anonymized, secure health data, enabling them to tap into the vast potential provided by the EU’s health data to inform scientific research, develop better treatments, and improve patient care.
EHR systems are classified as products under the EHDS Regulation and must comply with the essential requirements outlined in Annex II of the regulation. These requirements apply to the entire EHR system, not just individual software modules or harmonized components.
Manufacturers of EHR systems are responsible for ensuring CE marking and compliance. The CE marking confirms that the EHR system meets EU safety, performance, and interoperability standards. This includes developers of apps or platforms that allow patients or providers to access, store, or manage electronic health data in the priority categories (e.g. patient summaries, prescriptions, imaging, lab results).
Conclusion
The EU is continuously introducing new legislation that can affect medical devices and IVDs. These laws often have long transition periods, and manufacturers must stay informed to ensure future compliance. Manufacturers must consider all applicable legislation – not just MDR/IVDR – when declaring conformity. This includes both horizontal and harmonisation laws. In the next blog series, learn more about Medical Devices and Other EU Laws.
Related Next Steps
If you’re having difficulty navigating the EU regulatory landscape and struggling to comply with horizontal EU regulations, our experts specialise in helping manufacturers develop comprehensive regulatory strategies designed to ensure that medical devices and IVDs comply with all applicable EU legislation.
Contact us to discuss how we can support your efforts and stay tuned for more updates related to the European Medical Device landscape in our upcoming blog posts.
- Regulation (EU) No 1025/2012 on European Standardisation
- The word “Union” refers specifically to the European Union (EU) as a legal and political entity