




By Chris A. Parr – Principal, CRO
As a follow up to the “Recent developments in EU Horizontal Legislation” blog, we will now take a deeper dive into the multi-legislative compliance process for medical devices and in-vitro diagnostic medical devices (IVDs). Building on what we learned in the first instalment about product safety legislation and horizontal legislation, we will look at what it takes manufacturers to comply with sectorial, horizontal and national regulations.
Applicable Harmonisation (Sectorial) Legislation
Since the publication of the medical device regulation (MDR) and the IVD regulation (IVDR) in 2017 there has been a profound change in how medical devices and IVDs are regulated in the EU. Manufacturers are now well versed in the application of both regulations and their alignment with the new legislative framework and the market surveillance regulations1-3. Concepts such as economic operators, market surveillance, customs controls, and enforcement are significantly reinforced in the sectorial legislation and far better understood by industry.
In addition to the primary legislation, MDR and IVDR also introduced of a number of pieces of secondary legislation (implementing acts and delegated acts) that manufacturers should be aware of. Examples include the following:
- Regulation (EU) 2021/2226 on electronic instructions for use of medical devices
- Regulation (EU) 2022/2347 regards reclassification of groups of certain active products without an intended medical purpose
- Regulation (EU) 2023/2197 regards the assignment of Unique Device Identifiers for contact lenses
These implementing acts and delegated acts provide procedural and technical details to support the implementation of the MDR and IVDR. They should be used in conformity assessments whenever they are relevant.
Nationtal Legislation
While the MDR and IVDR are EU regulations and are directly applicable in all EU Member States, certain aspects require or allow for national implementation or additional national rules. National legislation can include provisions for the following:
- Clinical investigations
- Market Surveillance and Enforcement/fines
- Registration of Economic Operators
- Language and Labelling Requirements
Manufacturers must familiarise themselves with applicable national legislation and incorporate this into their market access strategies.
Relevant Horizontal Legislation
Manufacturers can however be misled into a false sense of security because of the underlying complexity associated with the application of EU legislation. CE marking a medical device or IVD often requires a hybrid approach which considers both the sector-specific harmonisation legislation and other horizontal legislation. Analogy can be drawn with standards where both sectorial (product standards) and horizontal standards (e.g. ISO 14971) may apply.
Generally speaking, the CE mark is required for products that address health, safety, environmental, and consumer protection requirements. In the EU there are currently thirty individual types of harmonisation legislation (sectorial) that are aligned with the new legislative framework approach. CE marking is mandatory for products covered by harmonisation legislation that explicitly require it. Some notable examples that have the potential to apply to medical devices include:
- RoHS – Directive 2011/65/EU
- Electromagnetic Compatibility – Directive 2014/30/EU
- Radio equipment – Directive 2014/53/EU
- Low Voltage – Directive 2014/35/EU
- Batteries – Regulation (EU) 2023/1542
- Machinery – Regulation (EU) 2023/1230
- Artificial Intelligence Act – Regulation (EU) 2024/1689
- Packaging and Packaging Waste – Regulation (EU) 2025/40
The are other EU regulations that are not harmonisation legislation per se but interact with harmonisation legislation. In other words, they do not require a CE mark in and of themselves in isolation. These are best understood as horizontal legislation that underpins and complements sectorial legislation. Horizontal rules refer to cross-sectorial regulations or guidelines that apply across multiple industries or policy areas, rather than being limited to a specific product or sector. These rules are designed to ensure consistency, coherence, and fairness in the application of EU law. Some notable examples that have the potential to apply to medical devices include:
- REACH Regulation – Regulation (EC) No 1907/2006
- CLP Regulation (EC) No 1272/2008
- Product Liability Directive – Directive 85/374/EEC / Directive (EU) 2024/2853
- General Data Protection Regulation (GDPR) – Regulation (EU) 2016/679
In most cases, it is almost impossible for a medical device to be CE marked solely based on compliance with the medical device or IVD sectorial legislation alone. There are multiple other sectorial regulations and horizontal regulations that may apply in addition to the MDR or the IVDR. All applicable EU legislation should be applied to a device when it is CE marked and captured in a single declaration of conformity. Therefore, CE marking is more than just an exercise in complying with the MDR or IVDR.
Medical device software is one possible exception because it can be delivered and consumed electronically and is essentially a digital asset. Therefore, there is a lower potential for other regulations to apply compared to physical devices. However, even software is subject to the product liability directive as far as defective products and financial coverage are concerned. Other EU rules related to artificial intelligence and cybersecurity may also apply to software.
Implications for QMS Design
Manufacturers need to take proactive steps to ensure that their quality management systems are designed to comply with all applicable regulations relating to products. In the past manufacturers have typically designed their QMSs specifically to comply with the requirements of ISO 13485, MDSAP and the medical device regulations in the EU and USA. However, increasingly it is necessary to design the QMS more broadly so that it considers aspects for all applicable regulations. For example, does the QMS require the consideration of other regulations as sources of design input or labelling.
Example: Active Implantable Device
Let us look at this through an example:
A manufacturer of a fictitious active implantable device. The device contains a battery, electrical circuits/components, software/firmware and can use radio frequency communications to communicate with a controller or programmer. In this example the medical device sectorial legislation is the primary legislation governing the CE marking of the device. And in this case a Notified Body designated under the MDR will complete the conformity assessment.
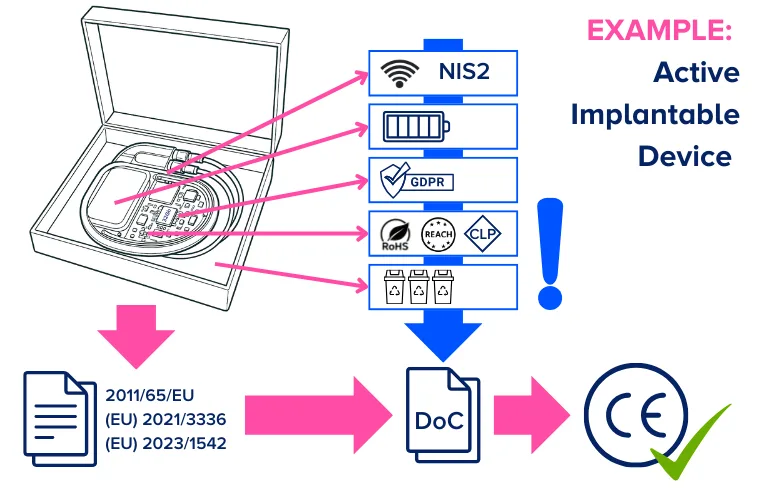
However, the device will also fall under the requirements of the RoHS directive (for the materials used in the electrical circuits/components), the radio equipment directive (for the communications), the NIS 2 Directive (for cybersecurity), the batteries regulation (for the battery) and the packaging regulation (for the packaging). In addition, the materials will need to be assessed in line with the REACH and CLP regulations. If the device collects, stores, transmits, or otherwise processes personal data GDPR applies.
In this case the device can only be CE marked when the applicable requirements of all the aforementioned directives and regulations have been fulfilled, and a single declaration of conformity drawn up. This may necessitate additional technical documentation and objective evidence for compliance. While the Notified Body is primarily focused on the review of the objective evidence related to compliance with the MDR, they can equally ask about the evidence to support other regulations.
New and Changing Regulations
There remains an ever-increasing production line of new and changing regulations in the EU that have the potential to affect medical devices. Often these are outside the core competencies of traditional medical device companies and their R&D, QA, and RA departments. Companies who can adapt, upskill, and move with the times will undoubtedly have the competitive advantage in the future when it comes to compliance and market access. The key enabler is building a management system that is constructed based on all applicable regulations and not just the narrow sectorial legislation for medical devices which has historically been the case. Also, manufacturers who can develop a holistic view of EU regulations and make it part of their regulatory intelligence process will be set up for future success. See our related blog on this topic.
Summary
The process to CE mark a medical device or IVD requires a comprehensive evaluation of all applicable harmonisation legislation, horizontal regulations and member state national legislation. CE marking based on the medical device regulation or IVD regulation alone is seldom sufficient to achieve a fully compliant and marketable device. Manufacturers must develop comprehensive and complete market access strategies to address all applicable requirements. In the context of the broader EU legal framework, the MDR and IVDR present several complexities and pain points that go beyond the regulations themselves. These challenges stem from how MDR and IVDR interact with other EU laws, institutions, and market dynamics. This multi-layered compliance increases complexity, especially for SMEs that lack regulatory resources.
Related next steps
If you’re having difficulty navigating the EU regulatory landscape and struggling to comply with horizontal EU regulations, our experts specialise in helping manufacturers develop comprehensive regulatory strategies designed to ensure that medical devices and IVDs comply with all applicable EU legislation.
Contact us to discuss how we can support your efforts and stay tuned for more updates related to the European Medical Device landscape in our upcoming blog posts.
- Regulation (EC) 765/2008 setting out the requirements for accreditation and the market surveillance of products
- Decision 768/2008 on a common framework for the marketing of products, which includes reference provisions to incorporate in product legislation revisions. In effect, it is a template for future product harmonisation legislation
- Regulation (EU) 2019/1020 on market surveillance and compliance of products
Further reading – Opportunity & Risk: Why we should open our minds to off-label use