




Why Real-World Evidence Matters
Regulators are increasingly open to real-world evidence (RWE) as a foundation for market entry, indication expansion, and post-market surveillance. Both the FDA and EU regulators recognize that randomized controlled trials can be overly burdensome and alone cannot capture the realities of device use in everyday clinical practice. Real-world evidence offers manufacturers a path to demonstrate ongoing safety, performance, and value without defaulting to costly or impractical studies. The opportunity is clear, but leveraging real world evidence effectively requires a strong grasp of the data sources, their strengths, and their limitations.
Defining the Basics: RWD vs. RWE
Real-world data (RWD) is information on patient health or healthcare delivery that is routinely collected in clinical practice. This includes data from medical records, claims, registries, surveys, and digitally connected devices. Real-world evidence (RWE) is what happens when this data is systematically analyzed to evaluate safety, performance, and other outcomes of interest. The distinction is subtle yet important. Data alone is not sufficient—its transformation into evidence is what regulators use to support decisions.
Where RWE Fits in Regulatory Strategy
RWE is most commonly applied to confirm safety and performance in the post-market phase as it is being used in standard care. Its potential, however, extends much further. It can support the expansion of indications when physicians pioneer new uses in practice, provided manufacturers are prepared to capture and analyze that data. It can also be applied to initial submissions in new regions, as long as differences in demographics, clinical practice, and standards of care are recognized and justified. In addition, RWE drawn from equivalent or similar devices can be used to benchmark performance or strengthen the design of a clinical investigation. The value is clear: RWE creates regulatory flexibility when applied strategically.
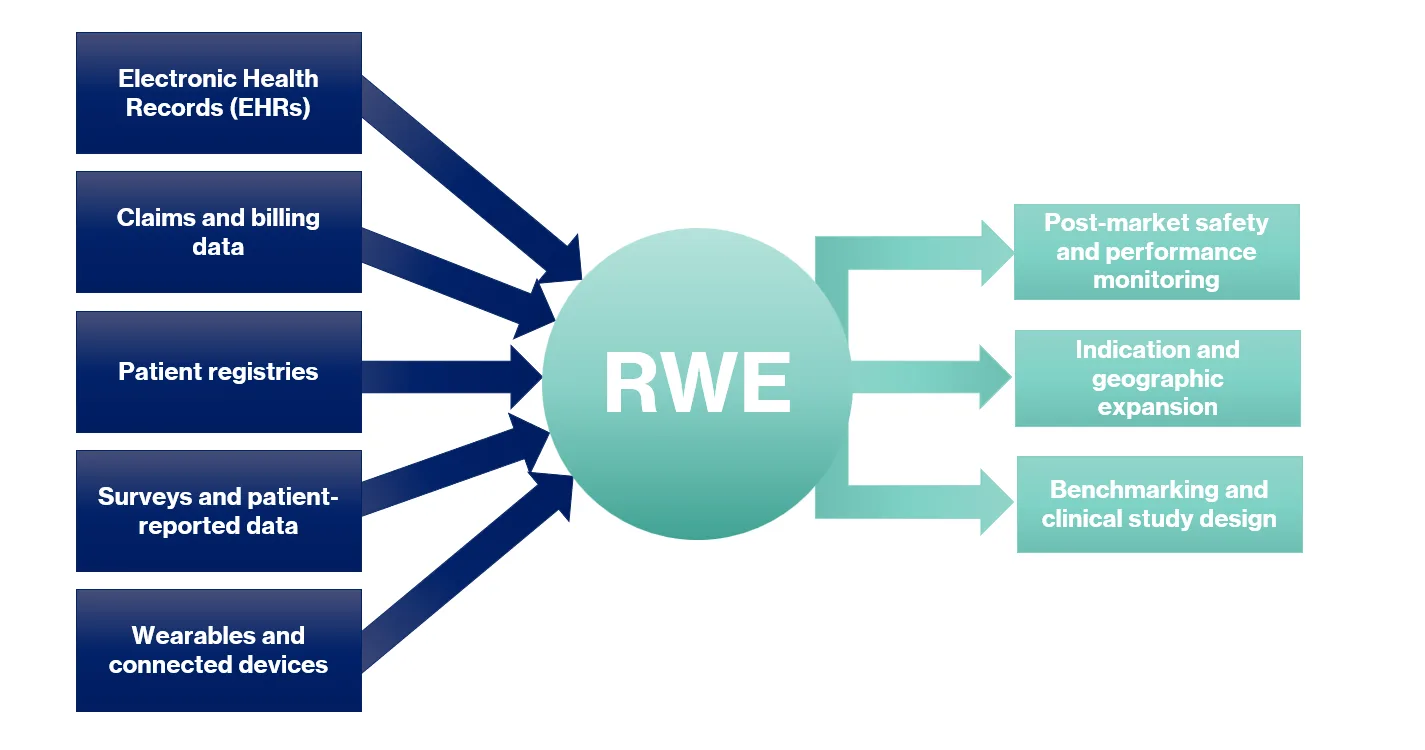
Methods
Use of RWE is not new to regulatory submissions. Historically, this evidence has been captured by retrospective chart reviews, registries, and claims data. Traditional retrospective chart reviews generate patient-level data but are limited to what is recorded during routine care, registries provide long-standing, regulator-accepted evidence but often lack device-specific linkage or complete follow-up and claims data, offers large structured datasets but rarely captures outcomes. More innovative approaches, such as survey-based chart reviews, allow healthcare providers to supply patient-level insights alongside reports of their own experience with the device. Comprehensive electronic health record datasets allow sponsors to see longitudinal outcomes across integrated systems, though linkage and access to unstructured notes remain challenges. Finally, the growing presence of wearables and connected devices allow for the creation of continuous streams of information, including patient-reported outcomes, that manufacturers can leverage with the proper consent.
Regulatory Considerations
It is important to recognize that RWE is not exempt from the rigor expected of clinical studies. Ethics approvals, formal protocols, and appropriate risk assessments are still required. The difference lies in the risk profile. Because RWE is observational and does not interfere with patient care, the main risk is disclosure of sensitive patient information.
Regulators also expect statistically sound sample sizes. The justification should be based on a defined objective and acceptance criterion for the primary endpoint. Submissions must be backed by robust design and statistical rationale to avoid rejection or deficiency findings.
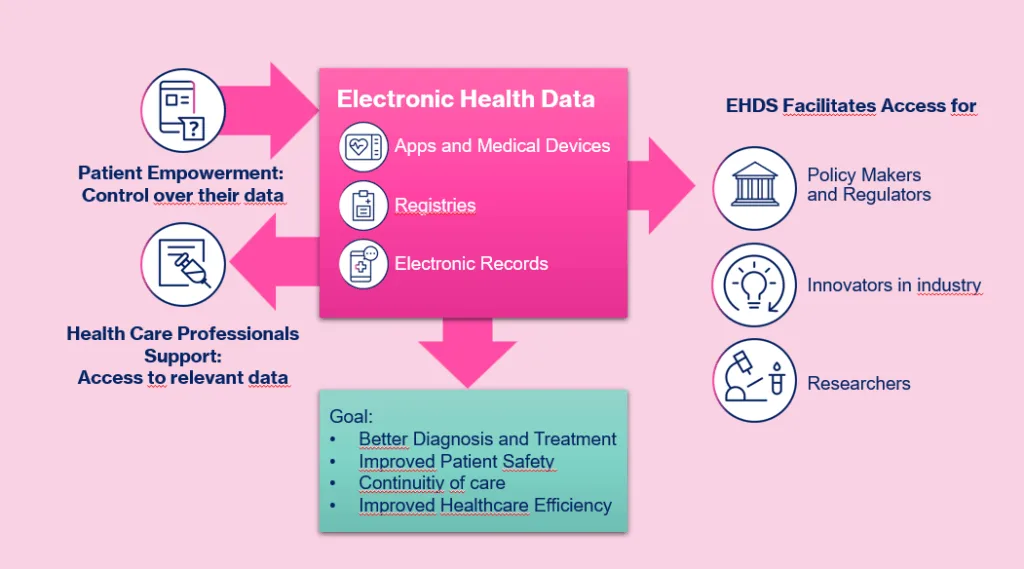
Emerging Frameworks: The EU Health Data Space
The European Health Data Space has the potential to greatly expand the accessibility of RWD. While its primary aim is to ensure continuity of care across the EU, it also designates industry as a beneficiary of secondary uses, including research and innovation. The practical details are still evolving, but the regulation signals a future where access to de-identified health data across borders may become more streamlined. For manufacturers, this could open new opportunities to build evidence more efficiently and across broader patient populations.
Practical Takeaways for Regulatory Teams
The key is to determine the need for generating clinical evidence, and to choose a method that is fit for purpose. There is no universal approach, and strategies must align with the device, the regulatory pathway, and the availability of data. Teams that embrace innovative methods like survey-based chart reviews or EHR datasets will be able to close evidence gaps faster and more efficiently than those relying solely on traditional investigational approaches. Justification remains central: whether bridging geographies, expanding indications, or using alternative comparators, regulators expect a clear and defensible rationale.
Engaging early with regulators is critical. Pre-submissions in the US or structured dialogues in the EU can clarify expectations and prevent missteps. Finally, teams should anticipate evolving access models. Frameworks like the EU Health Data Space will continue to reshape what is possible and what regulators are prepared to accept.
Key Take-Away
Real-world evidence is not a shortcut. It is a powerful complement to traditional studies when applied with precision. For MedTech teams, mastering the nuances of RWE collection and analysis can reduce costs, accelerate timelines, and strengthen submissions. Most importantly, it ensures that the regulatory strategy reflects the true performance of devices where it matters most: in the real world.
Exploring Outsourcing Clinical Trials in MedTech?
Full-service support isn’t just about doing more, it’s about designing stronger, more resilient product strategies from day one. Whether you’re bringing a first device to U.S. market or responding to shifting regulations, a partner like RQM+ brings the agility, depth, and cross-functional horsepower to accelerate progress with confidence. For MedTech leaders, the upside is clear: fewer delays, lower risk, and a faster path to clinical impact.
Prefer to start quickly? Begin with a consultative conversation.