



Slightly overdue, we wanted to highlight one of many discussion points in relation to proposals for international recognition pathways to access the Great Britain (GB) market. The recent update of the FDA’s data on Artificial Intelligence and Machine Learning (AI/ML)-Enabled Medical Devices has given us the opportunity to do just that.
Note: this is not intended to be an exhaustive look at how to get AI/ML-enabled medical devices onto the US, EU or UK markets.
The FDA’s List
On 7th August 2024, the U.S. Food and Administration (FDA) updated their list of Artificial Intelligence and Machine Learning (AI/ML)-Enabled Medical Devices, indicating that the FDA has authorized 950 AI/ML-enabled medical devices. The list is compiled based on publicly available information and all devices on the list have met the FDA’s applicable premarket requirements. The FDA are transparent that the list is not exhaustive, but the list does serve as a good indicator for this area of technology in the medtech industry.
Analysis of the FDA’s data shows a steady increase in the number of AI/ML-enabled medical devices over the last fourteen years (Figure 1). This increase is not surprising, given the general upturn in the use and availability of AI/ML-enabled products and services across all industries.

Another unsurprising element is the clinical areas in which the devices fall. Three quarters (76%) of the devices on the FDA’s list come under the Radiology panel (Figure 2), with the Cardiovascular panel taking 10% of devices on the list and 3rd place taken by the Neurology panel with only 4% of the devices. When looking at the Product Codes cited for each device on the FDA’s list, the top 10 Product Codes represent two-thirds of the list contents with LLZ and QIH taking nearly 30% of the overall total. Given the spread of devices by panel (Figure 2), there are no surprises that the top 10 codes all sit under the Radiology panel and are classified as Class 2 by the FDA (Table 1).
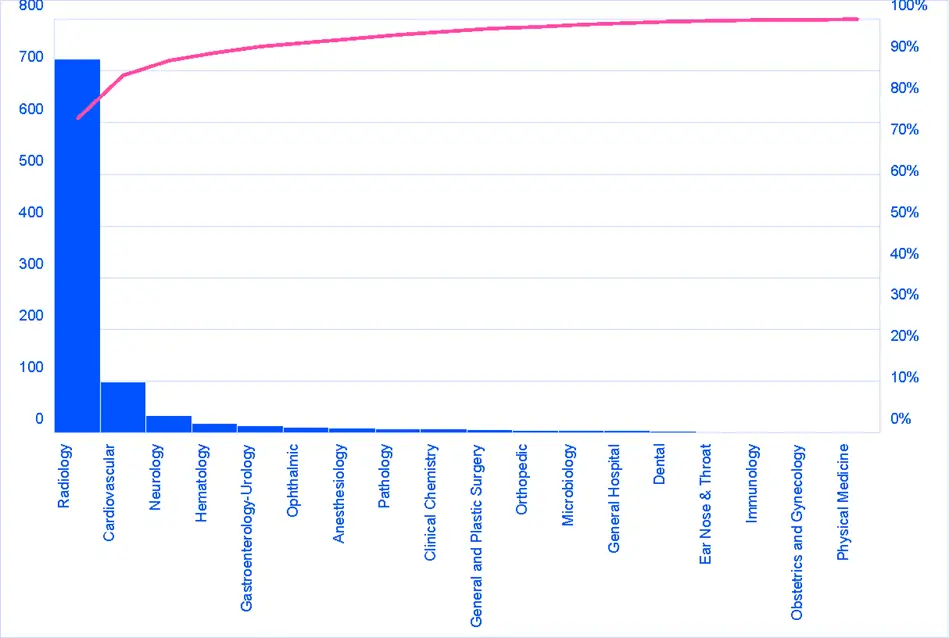
Table 1.Top 10 Product Codes from the FDA’s list
| Product Code | Device Name | Device Class | Regulation No. | TPLC Link |
|---|---|---|---|---|
| LLZ | system, image processing, radiological | 2 | 892.2050 | LLZ |
| QIH | automated radiological image processing software | 2 | 892.2050 | QIH |
| JAK | system, x-ray, tomography, computed | 2 | 892.1750 | JAK |
| IYN | system, imaging, pulsed doppler, ultrasonic | 2 | 892.1550 | IYN |
| LNH | system, nuclear magnetic resonance imaging | 2 | 892.1000 | LNH |
| QAS | radiological computer-assisted triage and notification software | 2 | 892.2080 | QAS |
| QFM | radiological computer-assisted prioritization software for lesion … | 2 | 892.2080 | QFM |
| QKB | radiological image processing software for radiation therapy | 2 | 892.2050 | QKB |
| MUJ | system, planning, radiation therapy treatment | 2 | 892.5050 | MUJ |
| MYN | analyzer, medical image | 2 | 892.2070 | MYN |
Digging further into the top 10 Product Codes on the FDA’s list (n=634, 66.7%), there was one device that went through the De Novo pathway (Product Code QAS) and two devices that went through the Premarket Approval (PMA) pathway (Product Code MYN). All of the rest of the devices (n=633) went through the 510(k) pathway, i.e. the subject device successfully demonstrated substantial equivalence with a predicate device (see BOX 1). As far as this author is aware, there is no current guidance from the FDA regarding how to compare the technical characteristics of AI/ML-enabled medical devices in order to demonstrate substantial equivalence. Examples of 510(k) summaries viewed (of devices on the FDA’s list) did compare inputs, user interfaces, image processing capabilities and operating systems but few revealed details of the training of the ML-model or its subsequent testing.
What is Substantial Equivalence?
A 510(k) requires demonstration of substantial equivalence to another legally U.S. marketed device. A device is substantially equivalent if, in comparison to a predicate it:
- has the same intended use as the predicate; and
- has the same technological characteristics as the predicate;
or
- has the same intended use as the predicate; and
- has different technological characteristics and does not raise different questions of safety and effectiveness; and
- the information submitted to FDA demonstrates that the device is as safe and effective as the legally marketed device.
The FDA’s guidance for the content of premarket submissions for device software functions does outline AI/ML specific considerations to be included in the device description, along with key information to be included in both the Basic and Enhanced documentation levels. There is however no clear discussion on how substantial equivalence needs to be demonstrated for an AI/ML-enabled medical device in terms of the technological characteristics, especially where the details of the training and test data are not always publicly available to enable a comparison of similarities and differences that could affect performance and safety.
It is a logical conclusion that these trends will continue in some form in the short to medium term. AI/ML-enabled devices will still predominantly be intended for radiological purposes and the majority will be incremental iterations on previous versions to enable access to the US market via the 510(k) pathway. Where is this going you may ask?
Global Collaboration and Harmonisation
The FDA have worked closely with Health Canada and the UK’s MHRA to develop good practice guidelines for AI/ML-enabled medical devices on the subjects of good machine learning practices, predetermined change control plans and transparency. These three regulatory authorities clearly share, at some level, a common vision on the best practices related to the increasing use of artificial intelligence, and more likely machine learning, in medical devices. But despite these previous collaborations and presumably continuing collaborations on this topic, there is a slight wrinkle to address; one that shines a light on the apparently different approaches to conformity assessments between two of those national competent authorities.
Crossing the Atlantic
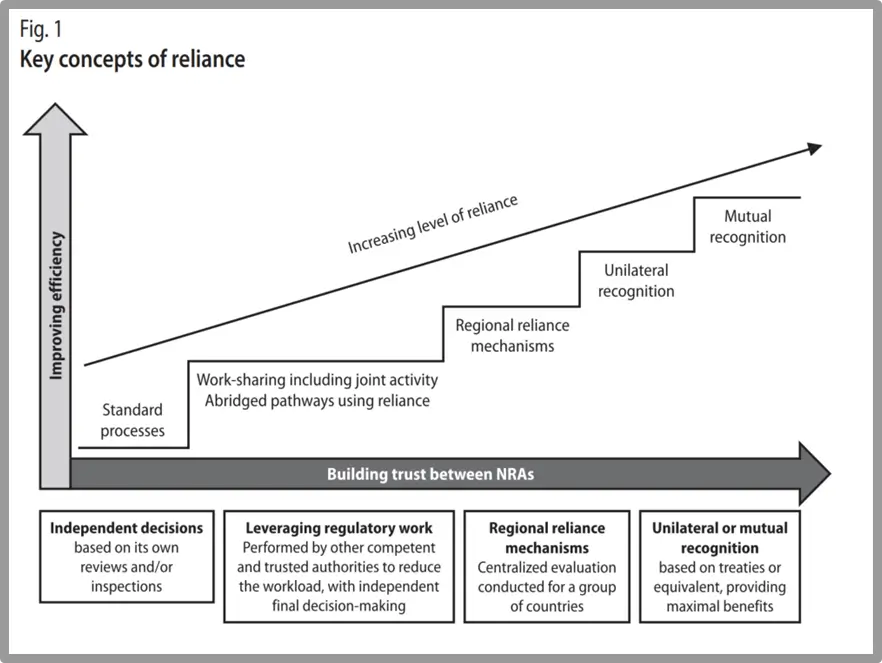
In May 2024 the MHRA announced their intentions for an international recognition policy for medical devices. This policy was published in the context of the MHRA’s work on the future regulatory framework for medical devices in the UK. The policy is built upon a combination of recognition (e.g. acceptance of a decision of another regulatory authority) and reliance (e.g. taking account of an assessment performed by another regulatory authority when performing your own assessment) (See Figure 3, and also WHO TRS 1033 Annex 10 for further information on good practices for reliance in regulation). Reliance and recognition can represent a ‘smarter’ approach to regulatory conformity assessments and market access, with the MHRA stating that reliance can result in “more predictable, faster approvals to improve access to quality-assured medical devices for patients”.
The MHRA’s statement of intent logically presents the use of reliance as a method to help mitigate resource constraints, by reducing duplication of effort in relation to performing conformity assessments for devices that have already been ‘approved’ for use by trusted regulatory authorities, and thus enable the MHRA’s resources to be directed towards more innovative products (see Box 2). To offer a level of control and reduce uncertainty, the MHRA, in their statement of intent, have identified four comparable regulator countries (CRCs), which includes the United States of America, specifically the FDA. Devices that have achieved market access in one or more of these countries/regions will be eligible for this proposed pathway into the GB market.
MHRA’s Consultation
This approach was part of the 2021 consultation by the MHRA, as part of the questions on Domestic Assurance. There were nearly 900 responses to the MHRA’s consultation in 2021, with just over 200 responses on the Domestic Assurance questions relating to MDSAP but only 66 responses to the specific question seeking opinion on whether the MHRA should allow routes to market that leverage approvals from other countries.
One may consider that AI/ML-enabled medical devices fall into innovative product territory. The FDA’s list says otherwise. Of the 950 devices on their list, 97% were cleared through the 510(k) pathway, meaning that they were substantially equivalent to a predicate. More incremental improvement than innovation some might say. A crude review of some of the 2024 clearance decisions (covering product codes LLZ and QIH, all under regulation 21 CFR 892.2050) showed that those clearances were based on multiple layers of equivalence (i.e. equivalent with a predicate that was equivalent with a predicate and so on).
In some cases, the predicates were previous generations of the manufacturer’s own device, in other cases the predicates were from various other manufacturers, and in some cases it was a mix of these two. It is this approach to equivalence (i.e. multiple layers of ‘piggybacking’ on a predicate), the potential for ‘product creep’ and the paucity of device-specific clinical data that is typically viewed with concern in the EU and the UK. This in part explains the stricter requirements around how to demonstrate equivalence in the EU MDR 2014/745 (compared to the requirements of the earlier Directives) and the MHRA’s stated intention to take the UK’s requirements for equivalence “beyond the equivalence requirements in the EU MDR” (UK Government response to consultation on the future regulation of medical devices in the United Kingdom, 2021).
In its current guise, the MHRA’s statement of intent has a list of nine exclusions, one of which is “SaMD (including AIaMD) products approved via a route which relies on equivalence to a predicate (US 510(k))”. That means the bulk of the FDA’s list of AI/ML-enabled medical devices are covered by this exclusion. For the purposes of this discussion there are two things that can be inferred from the MHRA’s list of exclusions:
- The MHRA are not fans of equivalence;
- Software as a medical device (SaMD), including AI/ML-enabled medical devices (MHRA refer to this as AIaMD) may be viewed more cautiously by the MHRA compared to the FDA.
So 924 of the AI/ML-enabled medical devices on the FDA’s list would be excluded from using the proposed international recognition pathway to access the Great Britain market. It’s not that they would fall into one of the three reliance pathways rather than the outright recognition pathway (which is only really available to lower risk devices such as Class I CE marked medical devices from the EU); but these 924 devices would be excluded from the whole recognition pathway and therefore would need to follow an alternative path to the GB (see examples in Box 3).
The MHRA’s exclusion specifically calls out the US 510(k), but it remains to be seen how the MHRA would view an AI/ML-enabled medical device that attained a CE mark by using equivalence as part of its clinical evidence strategy. More specifically, if the responsibility of the assessments for the four recognition and reliance pathways eventually sits with the UK Approved Bodies, how would the MHRA even know the strategies used by the device manufacturers to support the safety and performance of their device in order to obtain the CE mark.
Examples
Under the current proposal, manufacturers of AI/ML-enabled medical devices cleared for the US market via a 510(k) could access the GB market via one of the following routes:
i) UKCA conformity assessment process but there is currently a limited choice of UK Approved Bodies and uncertainty over when the future UK regulations will come into force and what they will contain, especially with regards to AI/ML-enabled medical devices.
ii) CE marking for the EU market, where safety and performance data for the subject device would be reviewed by a EU Notified Body, after which the device could then utilise the CE mark to access the GB market. The caveat here is the uncertainty of how Regulation 2024/1689 (AKA the AI Act) will be.
The proposed policy, and the exclusion of AI/ML-enabled devices cleared via the 510(k) path in the US, does not currently account for the different shades of 510(k) pathways, especially the requirement for special controls for specific product codes. These special controls can require very specific comparative testing and clinical data to be included within the 510(k) submission. Evidence packages of this nature, and the subsequent clearance based on that evidence, may be seen more favourably than the usual perception of devices cleared by claiming substantial equivalence to a predicate. Perhaps there could be scope for the exclusions to be modified with time. There is also no mention of the option for having stricter requirements for post-market surveillance (and subsequent reporting) as a proportionate measure that could enable these AI/ML-enabled medical devices to access the market via the recognition pathway but subsequently be monitored more closely once on the market.
Final thoughts
Medical devices that incorporate artificial intelligence or machine learning are on the rise. The data from the FDA indicates that the increase in these devices is not really a technological revolution, but more like quick-fire, iterative, evolution based upon existing devices. To push those devices down a more (at least in perception) burdensome conformity assessment route to access the GB market, may be an obstacle that device manufacturers choose to avoid.
It is possible that the current exploratory work on the international recognition policy may highlight some of these issues, or that the MHRA’s AI Airlock identifies elements of the market access pathway that could be improved. As much is made of regulatory harmonisation and alignment, it still seems that there are oddities to be resolved even between key collaborators in the harmonisation process.
Further Reading
- FDA Artificial Intelligence and Machine Learning (AI/ML)-Enabled Medical Devices
- FDA Artificial Intelligence and Machine Learning Software as a Medical Device Action Plan (January 2021)
- FDA Content of Premarket Submissions for Device Software Functions (14 June 2023)
- FDA Premarket Notification 510(k)
- FDA Proposed Regulatory Framework for Modifications to Artificial Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device (SaMD) – Discussion Paper and Request for Feedback (2 April 2019)
- FDA Software as a Medical Device (SAMD): Clinical Evaluation (adoption of IMDRF/SaMD WG/N41FINAL:2017) (8 December 2017)
- FDA Special Considerations for 510(k)s: Software
- FDA Technical Performance Assessment of Quantitative Imaging in Radiological Device Premarket Submissions (16 June 2022)
- FDA/MHRA/Health Canada Good Machine Learning Practice for Medical Device Development: Guiding Principles
- FDA/MHRA/Health Canada Predetermined Change Control Plans for Machine Learning-Enabled Medical Devices: Guiding Principles
- IMDRF consultation on Good machine learning practice for medical device development – Guiding Principles (Closed 30 Aug 2024)
- IMDRF consultation on Medical Device Software: Considerations for Device and Risk Characterization (Closed 2 May 2024)
- MDCG 2020-1 Guidance on clinical evaluation (MDR) / Performance evaluation (IVDR) of medical device software (March 2020)
- MHRA AI Airlock: the regulatory sandbox for AIaMD (last updated 21 August 2024)
- MHRA Crafting an intended purpose in the context of software as a medical device (SaMD) (22 March 2023)
- MHRA Medical device stand-alone software including apps (including IVDMDs) (v1.10f)
- MHRA’s Statement of policy intent: international recognition of medical devices
- Transparency for Machine Learning-Enabled Medical Devices: Guiding Principles
- WHO TRS 1033 Annex 10 Good reliance practices in the regulation of medical products.